
Abstract
Drug candidates identified by the pharmaceutical industry typically have unique structural characteristics to ensure they interact strongly and specifically with their biological targets. Identifying these characteristics is a key challenge for developing new drugs, and quantitative structure-activity relationship (QSAR) analysis has generally been used to perform this task. QSAR models with good predictive power improve the cost and time efficiencies invested in compound development. Generating these good models depends on how well differences between “active” and “inactive” compound groups can be conveyed to the model to be learned. Efforts to solve this difference issue have been made, including generating a “molecular descriptor” that compressively expresses the structural characteristics of compounds. From the same perspective, we succeeded in developing the Activity Differences-Quantitative Structure-Activity Relationship (ADis-QSAR) model by generating molecular descriptors that more explicitly convey features of the group through a pair system that performs direct connections between active and inactive groups. We used popular machine learning algorithms, such as Support Vector Machine, Random Forest, XGBoost and Multi-Layer Perceptron for model learning and evaluated the model using scores such as accuracy, area under curve, precision and specificity. The results showed that the Support Vector Machine performed better than the others. Notably, the ADis-QSAR model showed significant improvements in meaningful scores such as precision and specificity compared to the baseline model, even in datasets with dissimilar chemical spaces. This model reduces the risk of selecting false positive compounds, improving the efficiency of drug development.





Similar content being viewed by others
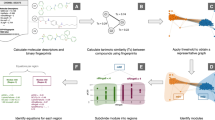
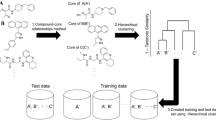
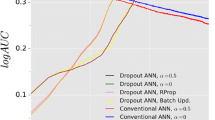
References
Mendez D, Gaulton A, Bento AP, Chambers J, De Veij M, Félix E, Magariños María P, Mosquera Juan F, Mutowo P, Nowotka M, Gordillo-Marañón M, Hunter F, Junco L, Mugumbate G, Rodriguez-Lopez M, Atkinson F, Bosc N, Radoux Chris J, Segura-Cabrera A, Hersey A, Leach Andrew R (2018) ChEMBL: towards direct deposition of bioassay data. J Nucleic Acids 47(D1):D930–D940. https://doi.org/10.1093/nar/gky1075
Zhu H (2020) Big data and artificial intelligence modeling for drug discovery. Annu Rev Pharmacol 60:573–589. https://doi.org/10.1146/annurev-pharmtox-010919-023324
Muhammad U, Uzairu A, Ebuka Arthur D (2018) Review on: quantitative structure activity relationship (QSAR) modeling. https://ijaar.org/articles/Volume4-Number5/Sciences-Technology-Engineering/ijaar-ste-v4n5-may18-p6.pdf. Accessed 19 Apr. 2018
Gedeck P, Kramer C, Ertl P (2010) Computational analysis of structure–activity relationships. Prog Med Chem 49:113–160. https://doi.org/10.1016/S0079-6468(10)49004-9
Xiong Y, Qiao Y, Kihara D, Zhang H-Y, Zhu X, Wei D-Q (2019) Survey of machine learning techniques for prediction of the isoform specificity of cytochrome P450 substrates. Curr Drug Metab 20(3):229–235. https://doi.org/10.2174/1389200219666181019094526
Seddon G, Lounnas V, McGuire R, van den Bergh T, Bywater RP, Oliveira L, Vriend G (2012) Drug design for ever, from hype to hope. J Comput Aided Mol Des 26(1):137–150. https://doi.org/10.1007/s10822-011-9519-9
Piir G, Kahn I, García-Sosa AT, Sild S, Ahte P, Maran U (2018) Best practices for QSAR model reporting: physical and chemical properties, ecotoxicity, environmental fate, human health, and toxicokinetics endpoints. Environ Health Perspect 126(12):126001. https://doi.org/10.1289/EHP3264
Reker D, Schneider G (2015) Active-learning strategies in computer-assisted drug discovery. Drug Discov Today 20(4):458–465. https://doi.org/10.1016/j.drudis.2014.12.004
Dearden JC (2017) The history and development of quantitative structure-activity relationships (QSARs). Oncology: breakthroughs in research and practice. IGI Global, UK. https://doi.org/10.4018/978-1-5225-0549-5.ch003
Livingstone DJ (2000) The characterization of chemical structures using molecular properties, a survey. J Chem Inf Comput 40(2):195–209. https://doi.org/10.1021/ci990162i
Hansch C, Fujita T (1964) p-σ-π analysis. A method for the correlation of biological activity and chemical structure. J Am Chem Soc 86(8):1616–1626. https://doi.org/10.1021/ja01062a035
Todeschini R, Consonni V (2008) Handbook of molecular descriptors. John Wiley & Sons, New York. https://doi.org/10.1002/9783527613106
Fujita T, Iwasa J, Hansch C (1964) A new substituent constant, π, derived from partition coefficients. J Am Chem Soc 86(23):5175–5180. https://doi.org/10.1021/ja01077a028
Ivanciuc O (2000) QSAR comparative study of Wiener descriptors for weighted molecular graphs. J Chem Inf Comput 40(6):1412–1422. https://doi.org/10.1021/ci000068y
Randić M (1991) Generalized molecular descriptors. J Math Chem 7(1):155–168. https://doi.org/10.1007/BF01200821
Durant JL, Leland BA, Henry DR, Nourse JG (2002) Reoptimization of MDL keys for use in drug discovery. J Chem Inf Comput 42(6):1273–1280. https://doi.org/10.1021/ci010132r
Rogers D, Hahn M (2010) Extended-connectivity fingerprints. J Chem Inf Model 50(5):742–754. https://doi.org/10.1021/ci100050t
Cramer RD, Patterson DE, Bunce JD (1988) Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins. J Am Chem Soc 110(18):5959–5967. https://doi.org/10.1021/ja00226a005
Ragno R (2019) www.3d-qsar. Com: a web portal that brings 3-D QSAR to all electronic devices—the Py-CoMFA web application as tool to build models from pre-aligned datasets. J Comput Aided Mol Des 33:855–864. https://doi.org/10.1007/s10822-019-00231-x
Pajor K (2020) Search for biological descriptors enabling artificial intelligence (AI) based quantified structure activity/relationship (QSAR/QSPR) models. https://ruj.uj.edu.pl/xmlui/handle/item/248823. Accessed 19 Apr. 2020
Muratov EN, Bajorath J, Sheridan RP, Tetko IV, Filimonov D, Poroikov V, Oprea TI, Baskin II, Varnek A, Roitberg A (2020) QSAR without borders. Chem Soc Rev 49(11):3525–3564. https://doi.org/10.1039/D0CS00098A
Xu J (2022) Evolving drug design methodology: from QSAR to AIDD. ChemRxiv. https://doi.org/10.26434/chemrxiv-2022-9fwmg
D’Souza S, Prema K, Balaji S (2020) Machine learning models for drug–target interactions: current knowledge and future directions. Drug Discov Today 25(4):748–756. https://doi.org/10.1016/j.drudis.2020.03.003
Vamathevan J, Clark D, Czodrowski P, Dunham I, Ferran E, Lee G, Li B, Madabhushi A, Shah P, Spitzer M (2019) Applications of machine learning in drug discovery and development. Nat Rev Drug Discov 18(6):463–477. https://doi.org/10.1038/s41573-019-0024-5
Siramshetty VB, Nguyen D-T, Martinez NJ, Southall NT, Simeonov A, Zakharov AV (2020) Critical assessment of artificial intelligence methods for prediction of hERG channel inhibition in the “big data” era. J Chem Inf Model 60(12):6007–6019. https://doi.org/10.1021/acs.jcim.0c00884
Gaulton A, Bellis LJ, Bento AP, Chambers J, Davies M, Hersey A, Light Y, McGlinchey S, Michalovich D, Al-Lazikani B (2012) ChEMBL: a large-scale bioactivity database for drug discovery. J Nucleic Acids 40(D1):D1100–D1107. https://doi.org/10.1093/nar/gkr777
Mysinger MM, Carchia M, Irwin JJ, Shoichet BK (2012) Directory of useful decoys, enhanced (DUD-E): better ligands and decoys for better benchmarking. J Med Chem 55(14):6582–6594. https://doi.org/10.1021/jm300687e
Landrum G (2013) RDKit: a software suite for cheminformatics, computational chemistry, and predictive modeling. http://www.rdkit.org/RDKit_Overview.pdf. Accessed 19 Apr. 2013
Rácz A, Bajusz D, Héberger K (2021) Effect of dataset size and train/test split ratios in QSAR/QSPR multiclass classification. Molecules 26(4):1111. https://doi.org/10.3390/molecules26041111
Datta S, Das S (2015) Near-Bayesian support vector machines for imbalanced data classification with equal or unequal misclassification costs. Int J Neural Netw 70:39–52. https://doi.org/10.1016/j.neunet.2015.06.005
Zhang L, Fourches D, Sedykh A, Zhu H, Golbraikh A, Ekins S, Clark J, Connelly MC, Sigal M, Hodges D, Guiguemde A, Guy RK, Tropsha A (2013) Discovery of novel antimalarial compounds enabled by QSAR-based virtual screening. J Chem Inf Model 53(2):475–492. https://doi.org/10.1021/ci300421n
Butina D (1999) Unsupervised data base clustering based on daylight’s fingerprint and tanimoto similarity: a fast and automated way to cluster small and large data sets. J Chem Inf Comput 39(4):747–750. https://doi.org/10.1021/ci9803381
Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V (2011) Scikit-learn: machine learning in python. https://www.jmlr.org/papers/volume12/pedregosa11a/pedregosa11a.pdf?ref=https:/. Accessed 19 Apr. 2011
Byvatov E, Schneider G (2003) Support vector machine applications in bioinformatics https://europepmc.org/article/med/15130823. Accessed 19 Apr. 2003
Svetnik V, Liaw A, Tong C, Culberson JC, Sheridan RP, Feuston BP (2003) Random forest: a classification and regression tool for compound classification and QSAR modeling. J Chem Inf Model 43(6):1947–1958. https://doi.org/10.1021/ci034160g
Sheridan RP, Wang WM, Liaw A, Ma J, Gifford EM (2016) Extreme gradient boosting as a method for quantitative structure–activity relationships. J Chem Inf Model 56(12):2353–2360. https://doi.org/10.1021/acs.jcim.6b00591
Chen T, Guestrin C (2016) Xgboost: a scalable tree boosting system. Proceedings of the 22nd acm sigkdd international conference on knowledge discovery and data mining. ArXiv.org 2016:785–794. https://doi.org/10.48550/arXiv.1603.02754
Berrar D (2019) Cross-Validation. In: Ranganathan S, Gribskov M, Nakai K, Schönbach C (eds) Encyclopedia of Bioinformatics and Computational Biology. Academic Press, Oxford. https://doi.org/10.1016/B978-0-12-809633-8.20349-X
Xu Y, Goodacre R (2018) On splitting training and validation set: a comparative study of cross-validation, bootstrap and systematic sampling for estimating the generalization performance of supervised learning. J Anal Test 2(3):249–262. https://doi.org/10.1007/s41664-018-0068-2
Agrawal T (2021) Hyperparameter optimization using scikit-learn. Hyperparameter optimization in machine learning. Springer, USA. https://doi.org/10.1007/978-1-4842-6579-6_2
Fawcett T (2006) An introduction to ROC analysis. Pattern Recognit Lett 27(8):861–874. https://doi.org/10.1016/j.patrec.2005.10.010
Stehman SV (1997) Selecting and interpreting measures of thematic classification accuracy. Remote Sens Lett 62(1):77–89. https://doi.org/10.1016/S0034-4257(97)00083-7
Huang J, Ling CX (2005) Using AUC and accuracy in evaluating learning algorithms. IEEE Trans Knowl Data 17(3):299–310. https://doi.org/10.1109/TKDE.2005.50
Baell JB, Holloway GA (2010) New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem 53(7):2719–2740. https://doi.org/10.1021/jm901137j
Senger MR, Fraga CA, Dantas RF, Silva FP Jr (2016) Filtering promiscuous compounds in early drug discovery: is it a good idea? Drug Discov Today 21(6):868–872. https://doi.org/10.1016/j.drudis.2016.02.004
Perkel JM (2015) Programming: pick up python. Nature 518(7537):125–126. https://doi.org/10.1038/518125a
Choi K-E, Balupuri A, Kang NS (2020) The study on the hERG blocker prediction using chemical fingerprint analysis. Molecules 25(11):2615. https://doi.org/10.3390/molecules25112615
Acknowledgements
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (NRF-2020R1A2C100691511).
Author information
Authors and Affiliations
Contributions
K.J.P. performed the calculations, analyzed the results, and wrote the manuscript. N.S.K.: designed, guided and approved the research work, rewrote the manuscript, and contributed to supervision, project administration, and funding acquisition. All authors have reviewed and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Park, G.J., Kang, N.S. ADis-QSAR: a machine learning model based on biological activity differences of compounds. J Comput Aided Mol Des 37, 435–451 (2023). https://doi.org/10.1007/s10822-023-00517-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10822-023-00517-1