
Abstract
The reaction of cobalt pivalate [Co(Piv)2]n and in situ generated N‑heterocyclic carbene IPrPh (1,3-bis(2,6-diisopropylphenyl)-2-phenylimidazol-4-ylidene) affords heteroligand complexes [Co2(Piv)4-(IPrPh)2] (I), [Co2(Piv)2.8(OtBu)1.2(IPrPh)2] (II), and [Co3(μ3-O)(Piv)4(IPrPh)2] (III). The structures of complexes II·C6H14 and III are determined by X-ray diffraction (XRD) (CIF files CCDC nos. 2216724 and 2216725, respectively). Exchange spin-spin interactions between the magnetic Со2+ ions in the synthesized compounds are estimated by quantum chemical calculations.
Similar content being viewed by others
INTRODUCTION
The organic ligands belonging to the class of N‑heterocyclic carbenes (NHC) have recently become a powerful tool that makes it possible to synthesize organometallic compounds of the very diverse structural types primarily possessing nontrivial catalytic properties [1, 2]. Just this feature of the carbene complexes predetermines a relevance of the development of new convenient methods for the synthesis of these compounds. It should be mentioned that the complexes of cobalt (in different oxidation states) with NHC are studied to a considerably less extent compared to the carbene derivatives of noble metals. However, among the known representatives of compounds of this type, the magnetically active cobalt complexes are characterized by very nontrivial geometries and electronic structures [3–8]. This leads to a high reactivity of the cobalt complexes with respect to small molecules [9, 10] and the appearance of a series of various useful functional properties [11–18]. In addition, the adducts of cobalt(II) imides with N-heterocyclic carbenes have the properties of single molecule magnets (SMM) and demonstrate one of the presently known highest reversal magnetization barriers among the 3d-metal complexes [19].
A specific feature of N-heterocyclic carbenes of the imidazoline series is a possibility of coordination to the complexing ion via two modes: via the normal (through the C(2) atom) and abnormal (through the C(4) atom) types (Scheme 1a, 1b). Abnormally coordinated carbenes are considered as stronger σ-donors, which can potentially affect substantially the magnetic properties of their complexes. The imidazole derivatives can be coordinated exclusively via the abnormal type upon the introduction of the aryl substituent into position 2 of the heterocycle [20]. Carbene with the phenyl substituent (IPrPh) was not obtained in the individual form, whereas the carbene with the biphenyl substituent (IPrBp) was isolated and characterized by XRD [21] (Scheme 1).
This work continues the cycle of studies on the development of synthetic approaches to the homo- and heterometallic carboxylate complexes with N‑heterocyclic carbenes [22–24] and is devoted to the search for methods of the synthesis of adducts of cobalt(II) pivalate with abnormally coordinated N‑heterocyclic carbene IPrPh. The electronic structures, geometric characteristics, and magnetic properties of the synthesized complexes were studied using quantum chemical simulation.

Scheme 1 .
EXPERIMENTAL
The synthesis was carried out in an inert atmosphere using the standard Schlenk technique. Solvents were dehydrated and degassed by boiling and distillation in an argon atmosphere using the corresponding drying agents [25]. Proligand IPrPh·HI was synthesized using a published procedure [20]. IR spectra in KBr pellets were recorded on a SCIMITAR FTS 2000 instrument. Elemental analysis was conducted at the Analytical Laboratory of the Nikolaev Institute of Inorganic Chemistry (Siberian Branch, Russian Academy of Sciences).
Synthesis of [Co2(Piv)4(IPrPh)2] (I). A mixture of [Co(Piv)2]n (95 mg, 0.364 mmol), IPrPh·HI (216 mg, 0.364 mmol), and KN(SiMe3)2 (75 mg, 0.357 mmol) was placed in a Schlenk tube, and THF (~20 mL) was condensed to the tube under reduced pressure using cooling with liquid nitrogen. After the spontaneous warming of the mixture from –196°C to room temperature, the resulting blue solution was heated at 60°C for 18 h, cooled, and evaporated to dryness. The solid residue was extracted with hexane (15 mL), and the blue extract was filtered through a glass filter (G4) and sealed in an evacuated L-shaped ampule. Dark blue crystals of I·C6H14 were formed after the slow evaporation of the solvent to the empty section of the ampule. (The standard crystallization procedure in a L-shaped ampule was described in detail [26] and demonstrated clearly in the supplemental material to the paper.) The yield was 90 mg (34%).
IR (KBr; ν, cm–1): 3086 w, 2963 s, 2929 m, 2870 m, 1606 s, 1574 s, 1481 m, 1469 m, 1418 m, 1356 m, 1223 m, 1133 m, 1058 w, 915 m, 891 m, 837 m, 778 m, 760 m, 718 w, 691 m, 609 m.
For C86H116N4O8Co2 | |||
Anal. calcd., % | C, 71.15 | H, 8.05 | N, 3.86 |
Found, % | C, 70.85 | H, 8.00 | N, 3.65 |
Synthesis of [Co2(Piv)2.8(OtBu)1.2(IPrPh)2] (II). A mixture of [Co(Piv)2]n (132 mg, 0.505 mmol), IPrPh· HI (302 mg, 0.509 mmol), and KOtBu (62 mg, 0.521 mmol) was placed in a Schlenk tube, and THF (~20 mL) was condensed to the vessel under reduced pressure using cooling with liquid nitrogen. After the spontaneous warming of the mixture from –196°C to room temperature, the resulting blue solution was heated at 60°C for 18 h, cooled, and evaporated to dryness. The residue was extracted with hexane (15 mL), and the blue extract was filtered through a glass filter (G4) and sealed in an evacuated L-shaped ampule. Dark blue crystals of II·C6H14 suitable for XRD were formed after the slow evaporation of the solvent into the empty section of the ampule. The yield was 120 mg (32%).
IR (KBr; ν, cm–1): 3083 m, 2964 s, 2929 m, 2871 m, 1610 s, 1597 s, 1574 s, 1492 m, 1480 m, 1469 m, 1417 m, 1395 m, 1354 m, 1255 w, 1221 m, 1104 m, 1059 w, 935 m, 887 m, 803 m, 778 m, 761 m, 717 w, 692 m, 608 m, 554 w.
For C90.8H130N4O6.8Co2 | |||
Anal. calcd., % | C, 72.50 | H, 8.71 | N, 3.72 |
Found, % | C, 72.85 | H, 8.50 | N, 3.55 |
Synthesis of [Co3(μ3-O)(Piv)4(IPrPh)2] (III). A mixture of [Co(Piv)2]n (88 mg, 0.337 mmol), IPrPh· HI (200 mg, 0.337 mmol), and KH (15 mg, 0.374 mmol) was placed in a Schlenk tube, and Et2O (~15 mL) was condensed into the vessel under a reduced pressure using cooling with liquid nitrogen. After the spontaneous warming of the mixture, the formed suspension was stirred at room temperature for 7 days until the end of gas evolution. The solution was filtered through a glass filter (G4) and sealed in a L-shaped ampule. Dark blue crystals of compound III suitable for XRD were formed after the slow evaporation of the solvent in the empty section of the ampule. The yield was 45 mg (26%).
IR (KB; ν, cm–1): 2964 s, 2929 m, 2870 m, 1560 s, 1481 m, 1465 m, 1459 m, 1414 m, 1400 m, 1356 m, 1222 m, 1059 w, 887 w, 803 w, 780 w, 761 w, 720 w, 695 w, 608 m, 597 w.
For C86H116N4OCo3 | |||
Anal. calcd., % | C, 67.66 | H, 7.66 | N, 3.67 |
Found, % | C, 67.45 | H, 7.35 | N, 3.70 |
XRD. All single-crystal structural studies of compounds II·C6H14 and III were carried out using a standard procedure on a Bruker D8 Venture automated diffractometer equipped with a CMOS PHOTON III detector and an IµS 3.0 microfocus source (MoKα radiation, λ = 0.71073 Å, focusing Montel mirrors). Reflection intensities were measured by the ω scanning of narrow (0.5°) frames. The data were reduced using the Apex3 software [27]. The structures were solved using the SHELXT program [28] and refined using the SHELXL program [29] in the anisotropic approximation for non-hydrogen atoms using the Olex2 program [30]. Hydrogen atoms were localized geometrically and refined in the rigid body approximation. The crystallographic characteristics of the complex and details of the diffraction experiment are given in Table 1.
Crystallographic data for II·C6H14 and III was deposited with the Cambridge Crystallographic Data Centre (CIF files CCDC nos. 2216724 and 2216725, respectively and are available at http://www. ccdc.cam.ac.uk/conts/retrieving.html).
Quantum chemical calculations. The calculations were performed using the Gaussian 16 program [31] by the density functional theory (DFT) method using the B3LYP functional [32] and Def2-SVP basis set. Dispersion interactions were taken into account applying the D3BJ empirical correction [33], the use of which makes it possible to correctly reproduce the geometric parameters obtained by XRD [34, 35]. Stationary points were localized on the potential energy surface (PES) by the full optimization of the geometries of the molecular structures with checking stability of the DFT wave function. Parameters of the exchange spin-spin interaction (J, cm–1) were calculated in terms of the broken symmetry (BS) formalism [36] using the Yamaguchi generalized spin projection method [37]. The graphical images of the optimized molecular structures were constructed using the ChemCraft program [38].
RESULTS AND DISCUSSION
Three approaches based on the interaction of the carbene precursor IPrPh·HI with bases KN(SiMe3)2, KOtBu, and KH were used to synthesize the cobalt pivalate complexes [Co(Piv)2]n with N-heterocyclic carbene IPrPh, since the target carbene is restrictedly stable. In the first case, the crystallization of the reaction product from hexane gave crystals of the [Co2(Piv)4(IPrPh)2]·C6H14 complex (I·C6H14). However, the quality of the isolated single crystals turned out to be very low and allowed us to determine the structural model only. The attempts to obtain qualitative single crystals from other solvents were unsuccessful. The file of crystallographic information containing the structural model of I·C6H14 was deposited with the Cambridge Structural Database (no. 2216723) and is not associated with this paper.

Scheme 2 .
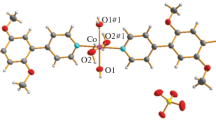
Complex [Co2(Piv)2.8(OtBu)1.2(IPrPh)2] (II) bearing different anionic ligands was isolated from the reaction of [Co(Piv)2]n, IPrPh·HI, and potassium tert-butylate. A molecule of complex II represents a centrosymmetric dimer in which two bridging positions are occupied by the pivalate and tert-butylate anions in a ratio of 2 : 3 (Fig. 1). The coordination sphere of the Co atoms is supplemented by the terminal κ1-coordinated pivalate anions and the IPrPh carbene molecules. The structure of compound II·C6H14 also contains hydrogen bonds between the CH groups of the carbene ligands and free O atoms of the terminal pivalate anions (C(21)···O(6) 3.156(7) Å, ∠C(21)H(21)O(6) 171.4°; C(54)···O(2) 3.217(8) Å, ∠C(54)H(54)O(2) 173.3°). The Co···Co distance is 3.223(3) Å.

Molecular structure of complex II. Thermal ellipsoids with 20% probability are shown; bridging pivalate anions and hydrogen atoms are omitted.
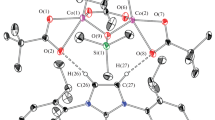
Trinuclear complex [Co3(μ3-O)(Piv)4(IPrPh)2] (III) was isolated from the reaction of [Co(Piv)2]n, IPrPh·HI, and potassium hydride. In the structure of complex III, three cobalt atoms are connected by the bridging μ3-O atom and three bridging pivalate anions binding in pairs the cobalt atoms (Fig. 2). The Co(1) and Co(2) atoms are additionally coordinated by the carbene ligands, whereas the Co(3) atom is additionally coordinated by the fourth pivalate anion. The Co–O distances in the fragment with the terminal pivalate anion (Co(3)–O(7) 1.951(4), Co(3)–O(8) 2.721(5) Å) and C–O bond lengths (1.286(7) and 1.238(7) Å) can hardly allow one to consider this anion as coordinated via the chelate mode. This conclusion is consistent with the results of DFT calculations that gave the model with a similar alternation of the Co–O and C–O bonds (Fig. 3). The Co–C distances in complex III (2.007(5) and 2.014(5) Å) are somewhat shorter than those in complex II but are close to the bond lengths for other coordination Co compounds with abnormally coordinated carbenes [39]. The sum of angles at the capped O(9) oxygen atom is 328.9°. The distances to the capped O atom in the Co3O fragment vary from 1.864(3) to 1.905(4) Å, and the shortest bond lengths correspond to the cobalt atoms with the coordinated carbene ligand. The structure of complex III also contains a weak hydrogen bond between the CH group of one of the carbene ligands and the free O atom of the terminal pivalate anion (C(22)···O(8) 3.413(7) Å, ∠C(22)H(22)O(8) 173.1°).

Molecular structure of complex III. Thermal ellipsoids with 30% probability are shown; hydrogen atoms are omitted.

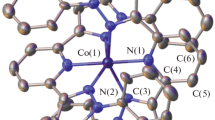
Optimized geometries of complexes II and III calculated by the B3LYP/Def2-SVP + D3BJ method. Hydrogen atoms are omitted; bond lengths are given in Å.
According to the quantum chemical calculation results (Table 2, Fig. 3), in considered complexes II and III, the Co2+ ions exist in the high-spin state, which is indicated by the values of the spin density on the metal centers (\(q_{{\text{s}}}^{{\text{M}}}\) ≈ 2.8). The geometry of the binuclear fragment and the Co···Co distance in complex II (being 3.21 Å) favor the appearance of a weak antiferromagnetic coupling of unpaired electron spins of the cobalt(II) ions (J = –11 cm–1). The exchange between the Co2+ ions enhances (J1 = –52 cm–1, J2 = –32 cm–1) on going to compound III. Possibly, its antiferromagnetic character is determined by both the presence of bridging groups with a high electron density delocalization on the Co–O–Co and Co–O–C–O–Co bridging fragments and the absence of a serious spin-orbital contribution.
Thus, we found new approaches to the synthesis of the heteroligand cobalt complexes with abnormal N‑heterocyclic carbene, whose composition is determined by efficient in situ carbene generation. In addition, the use of the DFT method for the simulation of the magnetic properties of the synthesized compounds made it possible to reveal antiferromagnetic channels of the spin-spin exchange between the paramagnetic cobalt(II) ions.
REFERENCES
N-Heterocyclic Carbenes: From Laboratory Curiosities to Efficient Synthetic Tools, Diez-Gonzalez, S., Ed., Croydon (UK): The Royal Society of Chemistry, 2016.
Hopkinson, M.N., Richter, C., Schedler, M., and Glorius, F., Nature, 2014, vol. 510, p. 485.
Wang, D., Leng, X., Ye, S., and Deng, L., J. Am. Chem. Soc., 2019, vol. 141, p. 7731.
Sinha, N., Pfund, B., Wegeberg, C., et al., J. Am. Chem. Soc., 2022, vol. 144, p. 9859.
Smith, J.M. and Long, J.R., Inorg. Chem., 2010, vol. 49, p. 11223.
Zolnhofer, E.M., Käß, M., Khusniyarov, M.M., et al., J. Am. Chem. Soc., 2014, vol. 136, p. 15072.
Massard, A., Braunstein, P., Danopoulos, A.A., et al., Organometallics, 2015, vol. 34, p. 2429.
Bellan, E.V., Poddel’sky, A.I., Protasenko, N.A., et al., ChemistrySelect, 2016, vol. 1, p. 2988.
Hu, X., Castro-Rodriguez, I., and Meyer, K., J. Am. Chem. Soc., 2004, vol. 126, p. 13464.
Gao, Y., Li, G., and Deng, L., J. Am. Chem. Soc., 2018, vol. 140, p. 2239.
Tokmic, K., Markus, C.R., Zhu, L., and Fout, A.R., J. Am. Chem. Soc., 2016, vol. 138, p. 11907.
Wang, D., Lai, Y., Wang, P., et al., J. Am. Chem. Soc., 2021, vol. 143, p. 12847.
Mo, Z., Xiao, J., Gao, Y., and Deng, L., J. Am. Chem. Soc., 2014, vol. 136, p. 17414.
Liu, Y. and Deng, L., J. Am. Chem. Soc., 2017, vol. 139, p. 1798.
Yu, R.P., Darmon, J.M., and Milsmann, C., J. Am. Chem. Soc., 2013, vol. 135, p. 13168.
Harris, C.F., Bayless, M.B., and van Leest, N.P., Inorg. Chem., 2017, vol. 56, p. 12421.
Deng, L., Bill, E., Wieghardt, K., and Holm, R.H., J. Am. Chem. Soc., 2009, vol. 131, p. 11213.
Tokmic, K., Greer, R.B., Zhu, L., and Fout, A.R., J. Am. Chem. Soc., 2018, vol. 140, p. 14844.
Yao, X.-N., Du, J.-Z., Zhang, Y.-Q., et al., J. Am. Chem. Soc., 2017, vol. 139, p. 37.
Ghadwal, R.S., Reichmann, S.O., and Herbst-Irmer, R., Chem.-Eur. J., 2015, vol. 21, p. 4247.
Rottschäfer, D., Glodde, T., Neumann, B., et al., Chem. Commun., 2020, vol. 56, p. 2027.
Nikolaevskii, S.A., Petrov, P.A., Sukhikh, T.S., et al., Inorg. Chim. Acta, 2020, vol. 508, p. 119643.
Yambulatov, D.S., Petrov, P.A., Nelyubina, Y.V., et al., Mendeleev Commun., 2020, vol. 30, p. 293.
Yambulatov, D.S., Nikolaevskii, S.A., and Shmelev, M.A., Mendeleev Commun., 2021, vol. 31, p. 624.
Gordon, A.J. and Ford, R.A., The Chemist’s Companion: A Handbook of Practical Data, Techniques, and References, New York: Wiley, 1972.
Petrov, P.A., Smolentsev, A.I., Bogomyakov, A.S., and Konchenko, S.N., Polyhedron, 2017, vol. 129, p. 60.
Apex3 Software Suite: Apex3, SADABS-2016/2, and SAINT. Version 2018.7-2, Madison: Bruker AXS Inc., 2017.
Sheldrick, G.M., Acta Crystallogr., Sect. A: Found. Adv., 2015, vol. 71, p. 3.
Sheldrick, G.M., Acta Crystallogr., Sect. C: Struct. Chem., 2015, vol. 71, p. 3.
Dolomanov, O.V., Bourhis, L.J., Gildea, R.J., et al., J. Appl. Crystallogr., 2009, vol. 42, p. 339.
Frisch, M.J., Trucks, G.W., Schlegel, H.B., et al., Gaussian 16. Revision C. 01, Wallingford: Gaussian, 2016.
Becke, A.D., J. Chem. Phys., 1992, vol. 96, p. 2155.
Grimme, S., Ehrlich, S., and Goerigk, L., J. Comput. Chem., 2011, vol. 32, p. 1456.
Chegerev, M.G., Piskunov, A.V., Tsys, K.V., et al., Eur. J. Inorg. Chem., 2019, vol. 2019, p. 875.
Tsys, K.V., Chegerev, M.G., Fukin, G.K., et al., Mendeleev Commun., 2020, vol. 30, p. 205.
Noodleman, L., J. Chem. Phys., 1981, vol. 74, p. 5737.
Shoji, M., Koizumi, K., Kitagawa, Y., et al., Chem. Phys. Lett., 2006, vol. 432, p. 343.
Chemcraft. Version 1.8. 2014. http://www.chemcraftprog.com.
Ghadwal, R.S., Lamm, J.-H., Rottschafer, D., et al., Dalton Trans., 2017, vol. 46, p. 7664.
Funding
This work was supported by the Russian Science Foundation, project no. 19-13-00436-P.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
The authors declare that they have no conflicts of interest.
Additional information
Translated by E. Yablonskaya
Rights and permissions
Open Access. This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Petrov, P.A., Nikolaevskii, S.A., Yambulatov, D.S. et al. Heteroleptic Cobalt Complexes with Abnormally Coordinated N-Heterocyclic Carbene. Russ J Coord Chem 49, 407–413 (2023). https://doi.org/10.1134/S1070328423600274
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1070328423600274