
Abstract
This paper presents a simple and rapid approach to the quantification of various glycosides using high-performance thin-layer chromatography (HPTLC). Different classes of glycosides, represented by genistin and ononin (both monosaccharidic O-glycosides), rutin (a disaccharidic O-glycoside) and luteolin-6-C-glucoside (a monosaccharidic C-glycoside), were successfully separated using a mixture of ethyl acetate‒methanol‒glacial acetic acid‒formic acid (11:1:1:1, V/V) as the mobile phase followed by derivatisation with natural product–polyethylene glycol (NP–PEG) reagent. The method was validated for the quantification of these glycosides in accordance with the guidelines of the International Council for Harmonisation. The general applicability of the validated approach is demonstrated with the analysis of a large number of glycosides including two glycosides (i.e. rutin, naringin) in commercial products.
Similar content being viewed by others

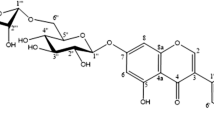
1 Introduction
A glycoside is an organic compound, usually of plant origin, comprising a sugar molecule linked to a non-sugar moiety referred to as aglycone or genin [1]. The most common bridging atom is oxygen, giving rise to O-glycosides, but can also be a sulphur (S-glycoside), nitrogen (N-glycoside) or carbon (C-glycoside) atom (Table 1) [2, 3]. The linkage between the sugar and the aglycone is a hemiacetal linkage formed by the reducing group of the sugar (usually aldehyde or ketone) and, in the case of O-glycosides, an alcoholic or phenolic hydroxyl group of the aglycon [1, 2]. In general, a distinction can be made between α-glycosides and β-glycosides, depending on the steric configuration of the hemiacetal hydroxyl group. The majority of the naturally occurring glycosides are β-glycosides [3, 4].
Generally, glycosides are more polar than their respective aglycones and, as a result, glycoside formation usually increases water solubility. Sometimes, the glycosidic residue is crucial for bioactivity, in other cases glycosylation only impacts on pharmacokinetic parameters [3,4,5], such as in vivo circulation, elimination and concentration in various body fluids [5, 6].
In plants, glycosides also play numerous important roles, such as converting toxic materials to non- or less toxic metabolites, protecting plants from bacteria and diseases through their antibacterial activity, regulation of growth, assisting in pollination and acting as a source of energy (i.e. sugar reservoir) [7, 8]. Many plants store chemicals in the form of inactive glycosides. These can be activated by enzymatic hydrolysis, which causes the sugar moiety to be removed, making the bioactive aglycone available for use [7,8,9]. There are also many glycosides in plants that are biologically active; however, their pharmacological effects are still largely determined by the structure of the respective aglycone [8, 9].
Glycosides are found in many important compound classes such as hormones, sweeteners, alkaloids, flavonoids and antibiotics [8,9,10]. Various medicines, condiments and dyes derived from plants exist as glycosides, for example, cardiac glycosides from Digitalis and Strophanthus, and antibiotics (e.g. streptomycin). Saponinic glycosides, on the other hand, can act as cleansing agents by lowering the surface tension of water [10, 11].
Current methods for the identification and quantification of glycosides are frequently based on high-performance liquid chromatography (HPLC), often coupled with another analytical technique [12,13,14,15], the most common are HPLC in tandem with diode array detection (HPLC‒DAD) or evaporative light scattering detection (HPLC‒ELSD). Another common technique is ultra-performance liquid chromatography coupled with mass spectrometry (UPLC‒MS/MS) [12,13,14,15].
High-performance thin-layer chromatography (HPTLC) is a sophisticated and increasingly popular tool for the analysis of complex natural products. It is a flexible and cost-efficient separation technique ideally suited for the analysis of constituents in botanicals and herbal drugs [16,17,18,19]. The advantages of full automation, scanning, selective detection principles, minimum sample preparation and hyphenation options, for example, have enabled HPTLC to be a powerful analytical tool to derive qualitative and quantitative information of compounds in complex mixtures presented by pharmaceuticals, natural products, and clinical and food samples [19,20,21,22,23].
The use of HPTLC has previously been reported for the analysis of some glycosides. For example, Chelyn et al. analysed various flavone C-glycosides (i.e. shaftoside, isoorientin, isovitexin, orientin, vitexinin) in the leaves of Clinacanthus nutans by both HPTLC and HPLC‒UV/DAD [24]. Another study conducted by Senguttuvan et al. identified pervoside and swertiamarin glycosides in Hypochaeris radicata via HPTLC [25], whereas Jaitak et al. developed and validated a HPTLC-based method for the quantification of three steviol glycosides (i.e. steviolbioside, stevioside and rebaudioside-A) in Stevia rebaudiana leaves [26]. The above mentioned studies are, however, only applicable to specific types of glycosides. There is still a need to develop a generally applicable HPTLC-based method that will be potentially suitable for the analysis of different types of glycosides.
This study reports on the development of such a novel HPTLC-based method to quantitatively analyse a wide range of glycosidic compounds. The selection of a suitable mobile phase is one of the critical factors in HPTLC analysis. The mobile phase development in this study referenced prior thin-layer chromatography (TLC)-based studies by Wagner et al. [27] and Jasprica et al. [28] for the analysis of flavonoid glycosides. However, their reported mobile phase, ethyl acetate‒water‒formic acid‒glacial acetic acid at a ratio of 100:26:11:11 (V/V), produced a fuzzy baseline under the operating conditions of the HPTLC, therefore necessitating further modifications. The final method adopted in this study was validated for accuracy, precision, linearity and sensitivity as per International Council for Harmonisation (ICH) guidelines.
2 Experimental
2.1 Chemicals and reagents
All reagents and solvents used were of analytical grade. Naringin was obtained from Alfa Aesar (Lancashire, UK), all other glycosides used in this study were sourced from ChemFaces (Wuhan, China). Methanol was purchased from Scharlau (Barcelona, Spain) and ethyl acetate, glacial acetic acid and formic acid from Ajax Finechem (Cheltenham, Australia). Silica gel 60 F254 HPTLC glass plates (20 × 10 cm) were purchased from Merck KGaA (Darmstadt, Germany). 2-Aminoethyl diphenylborinate was sourced from Chem Supply Australia Pty Ltd. (Port Adelaide, SA, Australia) and polyethylene glycol (PEG-400) was obtained from PharmAust Manufacturing (Welshpool, Western Australia).
2.2 Commercial samples
Commercial rutin capsules (450 mg rutin per capsule) were purchased from Now Foods (Bloomingdale, IL, USA). Naringin capsules (500 mg naringin per capsule) were sourced from Swanson Health Products (Fargo, ND, USA).
2.3 Reagent and sample preparation
A methanolic solution of 0.5 mg/mL naringin was prepared as a reference solution. Glycoside standards were prepared at a concentration of 20, 40, or 50 µg/mL in methanol (Table 1). Methanolic solutions of commercial rutin and naringin capsules were prepared at a concentration of 50 µg/mL. The natural product (NP) reagent [also known as Naturstoff reagent or Neu’s reagent, diphenylborinic acid 2-aminoethyl ester (DPBA) and 2-aminoethyl diphenylborinate] was used as the derivatising reagent as it is commonly applied for the detection of flavonoids and glycosides [29,30,31]. Natural product (NP)-derivatising reagent was prepared by dissolving 1 g of 2-aminoethyl diphenylborinate in methanol and then the volume of the solution was made up to 100 mL (1% m/V) [29]. Polyethylene glycol (PEG-400) reagent was prepared by mixing 5 g of polyethylene glycol in ethanol and then the volume of the solution was adjusted to 100 mL (5% m/V) [29].
2.4 Instrumentation and HPTLC method
2.4.1 Sample application
Four microlitres of the reference solution and specific volumes (Table 1) of the respective glycoside solutions were applied as 8 mm bands, 10 mm from the lower edge of the HPTLC plate at a rate of 150 nL s−1 using a semi-automated HPTLC application device (Linomat 5, CAMAG, Muttenz, Switzerland).
2.4.2 Development
The chromatographic separation was performed on silica gel 60 F254 HPTLC plates (glass plates 20 × 10 cm) in a saturated (33% relative humidity) automated development chamber (ADC2, CAMAG). The plates were pre-saturated with the mobile phase for 5 min, automatically developed to a distance of 70 mm at room temperature and dried for 5 min. The obtained chromatographic results were documented using a HPTLC imaging device (TLC Visualizer 2, CAMAG) at 254 nm. After derivatisation with NP–PEG reagent using a HPTLC derivatiser (3 mL NP reagent applied with green nozzle and 2 mL PEG reagent applied with blue nozzle), the images were visualised at R366 nm. The chromatographic images were digitally processed and analysed using specialised HPTLC software (visionCATS, CAMAG), which was also used to control the individual instrumentation modules.
Ethyl acetate, methanol, glacial acetic acid and formic acid at a ratio of 11:1:1:1 (V/V) were found to constitute a suitable mobile phase to separate the range of glycosides used in this study.
2.5 Method validation
The analytical method using the optimised mobile phase was validated for linearity, sensitivity, precision and accuracy according to ICH guidelines [32].
2.5.1 Linearity
Linearity refers to the ability of an analytical procedure to produce results in direct proportion to the concentration of an analyte within the defined concentration range [16, 17]. In this study, linearity was assessed by analysing four glycosidic test solutions (genistin, ononin, rutin and luteolin-6-C-glucoside) of various volumes (3‒15 μL) in a concentration range of 60‒300 ng/band. Three replicate measurements were conducted in three separate experiments for each sample. The HPTLC fingerprints of genistin, ononin, rutin and luteolin-6-C-glucoside are shown in Fig. 1. Peak heights were determined by the use of visionCATS for genistin and ononin prior to derivatisation and for rutin and luteolin-6-C-glycoside after derivatisation with NP-PEG. Linear five-point calibration curves for each glycoside were obtained and the obtained peak heights versus the corresponding concentrations of the samples evaluated by linear regression analysis. The coefficient of determination (r2), slope (m) and y-intercept (c) of the calibration curves were determined to assess the linearity of the developed method.

HPTLC image visualised at (a) 254 nm prior to derivatisation and (b) 366 nm after derivatisation with NP–PEG reagent: track 1: genistin; track 2: ononin; track 3: rutin; track 4: luteolin-6-C-glucoside; track 5: mixture of genistin, ononin, rutin and luteolin-6-C-glucoside
2.5.2 Sensitivity (limit of detection and limit of quantification)
Method sensitivity was assessed via the limit of detection (LOD) and limit of quantification (LOQ). LOD is defined as the lowest concentration at which the method is able to detect but not quantify a sample under the given experimental conditions [16, 17]. LOQ is the lowest amount of analyte that can be detected and quantified with suitable precision, accuracy and repeatability. The LOD and LOQ were calculated using the following equations:
where σ is the SD of the y-intercept and S is the average slope value of triplicate calibration curves.
2.5.3 Precision
Precision of a method is the degree of agreement among individual test results when the method is applied repeatedly to multiple samples. It characterises the closeness of results obtained from a series of measurements of a single sample analysed under the same conditions but at different times, on different instruments and/or by different operators. Precision can be considered at three levels, namely, repeatability (same operating conditions over a short interval of time), intermediate precision (within-laboratory variations, analysing the sample on different days, by different analysts and on different equipment) and reproducibility (between laboratories) [16, 17]. In this study, the method was validated for repeatability and intermediate inter-day precision by analysing in triplicate three different concentrations of the four respective glycoside test samples (80, 130 and 280 ng/band) and determining the percent relative standard deviation (%RSD) of the obtained peak height.
2.5.4 Accuracy
The accuracy of an analytical method is the degree of agreement of test results generated by the method with the true value [16, 17]. The accuracy of the method was determined through the percentage recovery of the four glycosidic test solutions analysed in triplicate at three different concentration levels (80, 130 and 280 ng/band).
3 Results and discussion
Specific volumes (3‒15 µL) of four glycoside solutions (20 µg/mL), namely genistin, ononin, rutin and luteolin-6-C-glucoside, were applied onto HPTLC plates followed by development using the optimised mobile phase. The HPTLC fingerprints of those glycosides are shown in Fig. 1. The specific glycosides are represented by bands of different colours (prior and after derivatisation with NP–PEG reagent) at RF 0.495 for genistin, RF 0.460 for ononin, RF 0.210 for rutin and RF 0.285 for luteolin-6-C-glucoside. The bands were well separated from each other, confirming the suitability of the method to analyse these glycosides with adequate specificity.
3.1 Linearity
Linearity was assessed by linear regression analysis of a five-point standard curve of the respective test glycosides over the concentration range of 60‒300 ng/spot. The coefficients of determination (r2) of three independent experiments are presented in Table 2. All the correlation coefficients had a value of > 0.99, suggesting adequate linearity of the HPTLC method in the defined concentration range.
3.2 Sensitivity (limit of detection and quantification)
The slope values and y intercepts derived from the linear regression analysis of the standard curves were used to calculate the LOD and LOQ for the four test glycosides (Table 2). The following LOD and LOQ values were found: 7.92 and 24.0 ng, respectively, for genistin; 7.76 and 23.52 ng, respectively, for ononin; 10.50 and 31.82 ng, respectively, for rutin; and 7.58 and 22.98 ng, respectively, for luteolin-6-C-glucoside.
3.3 Precision
The method was validated for repeatability and intermediate inter-day precision by analysing each of the four glycoside standards at three concentrations (80, 130 and 280 ng/spot) in 1 day and over 3 consecutive days. Precision was recorded as %RSD (Tables 3, 4). In both intra-day and inter-day precision, the %RSD was found to be less than 5% which is acceptable according to ICH guidelines, indicating that the method can be considered precise with high levels of confidence.
3.4 Accuracy
The accuracy of percentage mean recoveries was found to have less than 5% variation across all analyses for all four test glycosides (Table 5). The accuracy of percentage mean recoveries was found to range between 98.17% and 101.98% for genistin, 100.48% and 100.84% for ononin, 99.76% and 101.64% for rutin and between 99.73% and 100.30% for luteolin-6-C-glucoside (Table 3), variations that are all within the acceptable range of the ICH guideline.
3.5 Application of the validated method
3.5.1 Analysis of a wide range of glycoside standards
After its successful validation using the four test glycosides, the general applicability of the method for the quantification of a wider range of glycosides, representing four different categories of glycosides (Table 1), was demonstrated with the analysis of 15 additional glycoside standards. Their respective colour, RF, LOD and LOQ values are presented in Table 6. Their LOD and LOQ values ranged from 5.55 to 12.25 ng, and 18.30 to 34.55 ng, respectively. Sharp bands for the 15 glycoside standards were produced using the validated method (Figs. 2, 3). To derive a colour descriptor, the red, green and blue (RGB) values obtained for each band were converted into hue values H (°) and their corresponding colours (https://www.blog.jimdoty.com/?p=11507, accessed on 20 March 2023) [33]. It was found that upon derivatisation with NP–PEG reagent, all glycosides displayed distinct colours, ranging from cyan blue, orange and yellow–green to turquoise (Table 6). These colour descriptors might aid in compound identification. For example, hesperidin, naringin and luteolin-7-O-glucuronide have similar RF values (0.315, 0.325 and 0.330, respectively), which could hamper their identification, but present distinctly different colours upon derivatisation with NP reagent (cyan blue, turquoise and orange, respectively). The developed and validated method is demonstrated in this study to be capable of quantifying 15 glycosides without modifying the analytical technique which confirms the suitability of the approach to analyse a diverse range of glycosides.

HPTLC image at 254 nm of track 1: naringin; track 2: daidzin; track 3: calycosin-7-O-beta-glucoside; track 4: narcissoside; track 5: isovitexin; track 6: tiliroside; track 7: kaempferitrin; track 8: polytadin; track 9: quercitrin; track 10: isoquercetrin; track 11: hyperoside; track 12: phloridzin; track 13: sissotrin; track 14: luteolin-7-O-glucuronide; track 15: hesperidin

HPTLC image at 366 nm after derivatisation with NP–PEG of track 1: naringin; track 2: daidzin; track 3: calycosin-7-O-beta-glucoside; track 4: narcissoside; track 5: isovitexin; track 6: tiliroside; track 7: kaempferitrin; track 8: polytadin; track 9: quercitrin; track 10: isoquercetrin; track 11: hyperoside; track 12: phloridzin; track 13: sissotrin; track 14: luteolin-7-O-glucuronide; track 15: hesperidin
3.5.2 Analysis of commercial samples
To further demonstrate its usefulness and general applicability, the method was applied to the analysis of rutin and naringin in commercial samples. The commercial capsules containing rutin, which are marketed as a powerful combatant of free radicals, for the promotion of vascular health and the maintenance of the structural integrity of blood vessels, were produced from the flower buds of Sophora japonica. Naringin in the commercial sample was obtained from grapefruit peel (Citrus × paradisi) and is marketed as a glycosidic flavonoid which works synergistically with vitamin C to enhance cellular defence mechanisms. It was found that the analysis of rutin and naringin was not impacted by the presence of any excipient present in the capsules (Figs. 4, 5). An average (n = 3) of 448.30 mg of rutin per capsule was found in the commercial formulation stating to contain 450 mg per capsule (a percentage stated content of 99.62%), whereas commercial capsules with a stated content of 500 mg of naringin were found to contain an average of 496.80 mg (99.36%, n = 3) (Table 7). Thus, it can be concluded that the developed analysis method is capable of quantifying the content of the two glycosides in commercial samples (Figs. 6, 7).

HPTLC fingerprint of rutin at 366 nm derivatised with NP–PEG reagent. Tracks 1‒5: rutin standard (20 µg/mL, 3–15 µL); track 6: commercial rutin sample (50 µg/mL, 4 µL)

HPTLC fingerprint of naringin. Tracks 1–5: naringin standard (20 µg/mL, 8–16 µL); track 6: commercial naringin sample (50 µg/mL, 5 µL) at 366 nm derivatised with NP–PEG reagent

HPTLC software generated calibration curve for the quantification of rutin in commercial sample obtained after derivatisation with NP-PEG reagent at 366 nm

HPTLC software generated calibration curve for the quantification of naringin in commercial sample
Reviewing the literature, it appears that specific HPLC-based methods are limited in their applicability to the analysis of only certain types of glycosides [12, 33,34,35,36,37,38,39,40]. In HPLC analysis, substantial modifications (e.g. mobile phase, column, run time, flow rate) might be required for analysing different glycosidic groups [33, 38,39,40,41]. The current HPTLC-based method, on the other hand, is capable of analysing a wide range of glycosides efficiently using the same mobile phase, visualisation and derivatisation method. Many glycosides vary in their RF values which enables their individual identification and quantification. This was illustrated in this study with the analysis of genistin, ononin, rutin and luteolin-6-C-glucoside as test glycosides (Fig. 1) which produced distinctly different individual RF values (tracks 1‒4) even when analysed by HPTLC as a mixture (track 5).
Moreover, the validated method was successfully applied to the quantification of rutin and naringin in commercial capsules, where a stated content of more than 99% was confirmed for both samples, further illustrating the applicability of the method.
4 Conclusions
Glycosides play unique and vital roles in natural products. The growing field of molecular glycobiology, for instance, provides a clearer understanding of the relationship between aglycone and glycoside activity, which might help in developing more glycosidic drugs. These developments emphasise the importance of an accurate identification and quantification of a wide range of glycosides. There is scope for the development of new approaches to the quantitative analysis of glycosides as a complement or alternative to the current most commonly employed methods, including HPLC‒DAD analysis. HPTLC analysis offers such an approach as it is an easy, quick to perform and a powerful tool to analyse up to 15 samples of complex natural products or pharmaceuticals with minimal reagent input in a single run. In addition, HPTLC analysis generates multiple data sets, such as images taken at different light conditions (e.g. 254 and 366 nm, as well as at white light) before and after derivatisation, RF and RGB values of individual bands, their peak heights and peak areas as well as their respective ultraviolet (UV) and fluorescence spectra. This richness of data can set HPTLC analysis apart from other analytical techniques. This study has developed a simple HPTLC-based method suitable for the quantification of a wide range of glycosides, independent of their chemical nature (e.g. O-, C-glycosides, mono- and di-saccharide glycosides). The analytical approach was successfully validated for its specificity, linearity, sensitivity, precision and accuracy in accordance with ICH guidelines and its applicability for the quality control of herbal materials demonstrated with the quantification of glycosides in two commercial products.
References
Kytidou K, Artola M, Overkleeft HS, Aerts JMFG (2020) Plant glycosides and glycosidases: a treasure-trove for therapeutics. Front Plant Sci 11:357. https://doi.org/10.3389/fpls.2020.00357
Yao J, Weng Y, Dickey A, Wang YK (2015) Plants as factories for human pharmaceuticals: applications and challenges. Int J Mol Sci 16:28549–28565. https://doi.org/10.3390/ijms161226122
Zhang J, Wu J, Liu F, Tong L, Chen Z, Chen J, He H, Xu R, Ma Y, Huang C (2019) Neuroprotective effects of anthocyanins and its major component cyanidin-3-O-glucoside (C3G) in the central nervous system: an outlined review. Eur J Pharmacol 858:172500. https://doi.org/10.1016/j.ejphar.2019.172500
Xiao J, Capanoglu E, Jassbi AR, Miron A (2016) Advance on the flavonoid C-glycosides and health benefits. Crit Rev Food Sci Nutr 56:S29–S45. https://doi.org/10.1080/10408398.2015.1067595
Kren V, Martinkova L (2001) Glycosides in medicine: the role of glycosidic residue in biological activity. Curr Med Chem 8(11):1303–1328. https://doi.org/10.2174/0929867013372193
Wu L, Armstrong Z, Schröder SP, de Boer C, Artola M, Aerts JM, Overkleeft HS, Davies GJ (2019) An overview of activity-based probes for glycosidases. Curr Opin Chem Biol 53:25–36. https://doi.org/10.1016/j.cbpa.2019.05.030
Nyström L, Schär A, Lampi A (2012) Steryl glycosides and acylated steryl glycosides in plant foods reflect unique sterol patterns. Eur J Lipid Sci Technol 114:656–669. https://doi.org/10.1002/ejlt.201200033
Newman RA, Yang P, Pawlus AD, Block KI (2008) Cardiac glycosides as novel cancer therapeutic agents. Mol Interv 8:36–49. https://doi.org/10.1124/mi.8.1.8
Lin X, Ma L, Racette SB, Spearie CLA, Ostlund RE Jr (2009) Phytosterol glycosides reduce cholesterol absorption in humans. Am J Physiol Gastrointest Liver Physiol 296:G931–G935. https://doi.org/10.1152/ajpgi.00001.2009
Wink M (2015) Modes of action of herbal medicines and plant secondary metabolites. Medicines 2(3):251–286. https://doi.org/10.3390/medicines2030251
Kondo S, Hotta K (1999) Semisynthetic aminoglycoside antibiotics: development and enzymatic modifications. J Infect Chemother 5(1):1–9. https://doi.org/10.1007/s101560050001
Bylda C, Thiele R, Kobold U, Volmer DA (2015) Simultaneous quantification of digoxin, digitoxin, and their metabolites in serum using high performance liquid chromatography-tandem mass spectrometry. Drug Test Anal 7(10):937–946. https://doi.org/10.1002/dta.1781
Melo P, Machado R, Teixeira HM (2012) Analysis of digoxin and metildigoxin in whole blood using solid-phase extraction and liquid chromatography tandem mass spectrometry. Int J Anal Chem 2012:975824. https://doi.org/10.1155/2012/975824
Kaiser P, Akerboom T, Wood WG, Reinauer H (2006) A novel LC-IDMS/MS method for the determination of the cardiac glycosides digoxin and digitoxin using caesium adducts. Clin Lab 52(1–2):37–42
Vlase L, Popa DS, Muntean D, Mihu D, Leucuta S (2009) A new, high-throughput high-performance liquid chromatographic/mass spectrometric assay for therapeutic level monitoring of digoxin in human plasma. J AOAC Int 92(5):1390–1395
Patel KG, Patel VG, Patel KV, Gandhi TR (2010) Validated HPTLC method for quantitative D termination of Gallic acid in stem bark of Myrica esculenta Buch.-Ham. Ex D. Don, Myricaceae. J AOAC Int 93(5):1422–1427.
Wang J, Tang F, Yue Y, Guo X, Yao X (2010) Development and validation of an HPTLC method for simultaneous quantitation of isoorientin, isovitexin, orientin, and vitexin in bamboo-leaf flavonoids. J AOAC Int 93(5):1376–1383
Sudberg S, Sudberg EM, Terrazas J, Sudberg S, Patel K, Pineda J, Fine B (2010) Fingerprint analysis and the application of HPTLC to the determination of identity and quality of botanicals, from an industry perspective. J AOAC Int 93(5):1367–1375
Arup U, Ekman S, Lindblom L, Mattsson JE (1993) High performance thin layer chromatography (HPTLC), an improved technique for screening lichen substances. Lichenologist 25(1):61–71. https://doi.org/10.1006/lich.1993.1018
Sherma J (2010) Review of HPTLC in drug analysis: 1996–2009. J AOAC Int 93(3):754–764
Chetna K, Komal O, Vadia N (2022) Development of HPTLC method for the simultaneous estimation of quercetin, curcumin, and ascorbic acid in herbal formulations. J Iran Chem Soc 19(10):4129–4138. https://doi.org/10.1007/s13738-022-02586-9
Chaudhari BG, Patel NM, Shah PB, Modi KP (2006) Development and validation of a HPTLC method for the simultaneous estimation of atorvastatin calcium and ezetimibe. Indian J Pharm Sci 68(6):793–796. https://doi.org/10.4103/0250-474X.31018
Ristivojevic P, Trifkovic J, Vovk I, Milojkovic-Opsenica D (2017) Comparative study of different approaches for multivariate image analysis in HPTLC fingerprinting of natural products such as plant resin. Talanta 162:72–79. https://doi.org/10.1016/j.talanta.2016.10.023
Chelyn JL, Omar MH, Mohd Yousof NS, Ranggasamy R, Wasiman MI, Ismail Z (2014) Analysis of flavone C-glycosides in the leaves of Clinacanthus nutans (Burm. f.) Lindau by HPTLC and HPLC-UV/DAD. Sci World J 2014:724267. https://doi.org/10.1155/2014/724267
Senguttuvan J, Subramaniam P (2016) HPTLC fingerprints of various secondary metabolites in the traditional medicinal Herb Hypochaeris radicata L. J Bot 2016:5429625. https://doi.org/10.1155/2016/5429625
Jaitak V, Gupta AP, Kaul VK, Ahuja PS (2008) Validated high-performance thin-layer chromatography method for steviol glycosides in Stevia rebaudiana. J Pharm Biomed Anal 47(4–5):790–794. https://doi.org/10.1016/j.jpba.2008.03.022
Wagner H, Bladt S (1996) Plant drug analysis—a thin layer chromatography Atlas, 2nd edn, chap. 7. Springer, Berlin
Jasprica I, Smolčić-Bubalo A, Mornar A, Medić-Šarić M (2004) Investigation of the flavonoids in croatian propolis by thin-layer chromatography. J Planar Chromat-Mod TLC 17:95–101
Gafner S, Bergeron C, Batcha LL, Angerhofer CK, Sudberg S, Sudberg ÉM, Guinaudeau H, Gauthier R (2003) Analysis of Scutellaria lateriflora and Its adulterants Teucrium canadense and Teucrium chamaedrys by LC–UV/MS, TLC, and digital photomicroscopy. J AOAC Int 86:453–460
Reich E, Schibli A (2011) High-performance thin-layer chromatography for the analysis of medicinal plants. Thieme, New York
Jug U, Glavnik V, Kranjc E, Vovk I (2018) HPTLC–densitometric and HPTLC–MS methods for analysis of flavonoids. J Liq Chromatogr Relat Technol 41:329–341. https://doi.org/10.1080/10826076.2018.1448690
ICH Harmonized Tripartite Guideline: Validation of Analytical Procedures: Text and Methodology Q2(R1) Geneva, International Conference on Harmonization (2005).
Lawag IL, Sostaric T, Lim LY, Hammer K, Locher C (2022) The development and application of a HPTLC-derived database for the identification of phenolics in honey. Molecules 27(19):6651. https://doi.org/10.3390/molecules27196651
Albrecht HP (1999) Cardiac glycosides. In: Ikan R (ed) Naturally occurring glycosides. Wiley, Chichester, pp 83–123.
Maurya A, Manika N, Verma RK, Singh SC, Srivastava SK (2013) Simple and reliable methods for the determination of three steroidal glycosides in the eight species of Solanum by reversed-phase HPLC coupled with diode array detection. Phytochem Anal 24(1):87–92. https://doi.org/10.1002/pca.2387
Dinakaran SK, Chelle S, Avasarala H (2019) Profiling and determination of phenolic compounds in poly herbal formulations and their comparative evaluation. J Tradit Complement Med 9(4):319–327. https://doi.org/10.1016/j.jtcme.2017.12.001
Agatonovic-Kustrin S, Gegechkori V, Petrovich DS, Ilinichna KT, Morton DW (2021) HPTLC and FTIR fingerprinting of olive leaves extracts and ATR-FTIR characterisation of major flavonoids and polyphenolics. Molecules 26(22):6892. https://doi.org/10.3390/molecules26226892
Yao X, Zhou GS, Tang YP, Qian YF, Guan HL, Pang H, Zhu S, Mo X, Su SL, Jin C, Qin Y, Qian DW, Duan JA (2013) Simultaneous quantification of flavonol glycosides, terpene lactones, biflavones, proanthocyanidins, and ginkgolic acids in Ginkgo biloba leaves from fruit cultivars by ultrahigh-performance liquid chromatography coupled with triple quadrupole mass spectrometry. Biomed Res Int 2013:582591. https://doi.org/10.1155/2013/582591
Mishra S, Aeri V (2017) Biotransformation of lignan glycoside to its aglycone by Woodfordia fruticosa flowers: quantification of compounds using a validated HPTLC method. Pharm Biol 55(1):360–366. https://doi.org/10.1080/13880209.2016.1238948
Shanker K, Gupta MM, Santosh SK, Bawankule DU, Pal A, Khanuja SPS (2007) Determination of bioactive nitrile glycoside(s) in drumstick (Moringa oleifera) by reverse phase HPLC. Food Chem 105(1):376–382. https://doi.org/10.1016/j.foodchem.2006.12.034
Lampert EC, Bowers MD (2011) A comparison of sample preparation techniques for quantifying Iridoid glycosides sequestered by Lepidopteran larvae. J Chem Ecol 37(5):496–499. https://doi.org/10.1007/s10886-011-9941-4
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions. No funding was received for conducting this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no competing interests to declare that are relevant to the content of this article.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sultana, S., Foster, K., Hossain, M.L. et al. Development and validation of an assay for the quantification of glycosides using high-performance thin-layer chromatography (HPTLC). JPC-J Planar Chromat 36, 179–190 (2023). https://doi.org/10.1007/s00764-023-00239-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00764-023-00239-y