
Abstract
Background
Aminoacylase-1 deficiency (ACY1D) is an autosomal recessive rare inborn error of metabolism, which is caused by disease-causing variants in the ACY1. This disorder is characterized by increased urinary excretion of specific N-acetyl amino acids. Affected individuals demonstrate heterogeneous clinical manifestations which are primarily neurologic problems. In neuroimaging, corpus callosum hypoplasia, cerebellar vermis atrophy, and delayed myelination of cerebral white matter have been reported.
Aims
Finding disease-causing variant and expanding imaging findings in a patient with persistent basal ganglia involvement.
Methods
Whole-exome sequencing was performed in order to identify disease-causing variants in an affected 5-year-old male patient who presented with neurologic regression superimposed on neurodevelopmental delay following a febrile illness. He had inability to walk, cognitive impairment, speech delay, febrile-induced seizures, truncal hypotonia, moderate to severe generalized dystonia, and recurrent metabolic decompensation.
Results
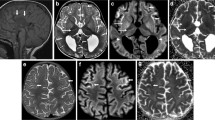
All metabolic tests were normal except for a moderate metabolic acidosis following febrile illnesses. The results of serial brain magnetic resonance imaging (MRI) at ages 1 and 4.5 years revealed persistent bilateral and symmetric abnormal signals in basal ganglia mainly caudate and globus pallidus nuclei with progression over time in addition to a mild supratentorial atrophy. A homozygous missense variant [NM_000666.3: c.1057C>T; p.(Arg353Cys)] was identified in the ACY1, consistent with aminoacylase-1 deficiency. Variant confirmation in patient and segregation analysis in his family were performed using Sanger sequencing.
Conclusions
Our findings expanded the phenotype spectrum of ACY1-related neurodegeneration by demonstrating persistent basal ganglia involvement and moderate to severe generalized dystonia.


Similar content being viewed by others


Availability of data and materials
Human variant and pertinent phenotypes have been reported to ClinVar (Submission ID: SUB9358917; Accession ID: SCV001547499).
References
Ferri L, Funghini S, Fioravanti A, Biondi E, la Marca G, Guerrini R, Donati MA, Morrone A (2014) Aminoacylase I deficiency due to ACY1 mRNA exon skipping. Clin Genet 86(4):367–372
Sass JO, Mohr V, Olbrich H, Engelke U, Horvath J, Fliegauf M, Loges NT, Krantz SS, Moebus R, Weiler P, Kispert A, Furga AS, Wevers RA, Omran H (2006) Mutations in ACY1, the gene encoding aminoacylase 1, cause a novel inborn error of metabolism. Am J Hum Genet 78(3):401–409.
Sass JO, Vaithilingam J, Gemperle-Britschgi C, Delnooz CC, Kluijtmans LA, van de Warrenburg BP, Wevers RA (2016) Expanding the phenotype in aminoacylase 1 (ACY1) deficiency: characterization of the molecular defect in a 63-year-old woman with generalized dystonia. Metab Brain Dis 31(3):587-592.
Alessandrì MG, Casarano M, Pezzini I et al (2014) Isolated mild intellectual disability expands the aminoacylase 1 phenotype spectrum. JIMD Rep Vol 16. Springer, pp 81–7
Lindner HA, Lunin VV, Alary A, Hecker R, Cygler M, Ménard R (2003) Essential roles of zinc ligation and enzyme dimerization for catalysis in the aminoacylase-1/M20 family. J Biol 278(45):44496–44504.
Van Coster RN, Gerlo EA, Giardina TG, Engelke UF, Smet JE, De Praeter CM, Meersschaut VA, De Meirleir LJ, Seneca SH, Devreese B, Leroy JG, Herga S, Perrier JP, Wevers RA, Lissens W (2005) Aminoacylase I deficiency: a novel inborn error of metabolism. Biochem Biophys Res Commun 338(3):1322–1326.
Sass J, Olbrich H, Mohr V, Hart C, Woldseth B, Krywawych S, Bjurulf B, Lakhani PK, Buchdahl RM, Omran H (2007) Neurological findings in aminoacylase 1 deficiency. Neurology 68(24):2151–2153.
Zaki OK, Krishnamoorthy N, El Abd HS, Harche SA, Mattar RA, Nofal MY, Bekay RE, Ahmed KA, Doss CGP, Zayed H (2017) Two patients with Canavan disease and structural modeling of a novel mutation. Metab Brain Dis 32(1):171–177.
Gemmill RM, Varella-Garcia M, Smith DI, Erickson P, Golembieski W, Miller Y, Coyle-Morris J, Tommerup N, Drabkin HA (1991) A 2.5-Mb physical map within 3p21. 1 spans the breakpoint associated with Greig cephalopolysyndactyly syndrome. Genomics 11(1):93–102.
Gaaib JN, Nassief AF, Al-Assi A (2011) Simple salting-out method for genomic DNA extraction from whole blood. Tikrit J Pure Sci 16(2):1813–1662.
Mohammadi P, Daneshmand MA, Mahdieh N, Ashrafi MR, Heidari M, Garshasbi M (2021) Identification of a novel missense c. 386G> A variant in a boy with the POMGNT1-related muscular dystrophy-dystroglycanopathy. Acta Neurol Belg 121(1):143–151.
Tavasoli AR, Memar EHE, Ashrafi MR, Hosseini SMM, Haghighi R, Ghabeli H, Pourbakhtyaran E, Rasoulinezhad M, Mohammadi P, Heidari M (2022) Primary and secondary microcephaly, global developmental delay, and seizure in two siblings caused by a novel missense variant in the ZNF335 gene. J Mol Neurosci 72(4):719–729.
Ashrafi MR, Haghighi R, Badv RS, Ghabeli H, Tavasoli AR, Pourbakhtyaran E, Rezaei Z, Mahdieh N, Mohammadi P, Morteza M 2022) Epilepsia partialis continua a clinical feature of a missense variant in the ADCK3 gene and poor response to therapy. J Mol Neurosci 72(5):1125–1132.
Yuliani D, Jannah A (eds) (2017) Primer design and in silico analysis of endoglucanase gene for Bacillus genus. 1st International Conference in One Health (ICOH 2017). Atlantis Press
Rosenbloom KR, Armstrong J, Barber GP, Casper J, Clawson H, Diekhans M, et al (2015) The UCSC genome browser database: 2015 update. Nucleic Acids Res 43(D1):D670–D681.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17(5):405–423.
Thorvaldsdóttir H, Robinson JT, Mesirov JP (2013) Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief Bioinformatics 14(2):178–192
Rebowski G, Boczkowska M, Drazic A, Ree R, Goris M, Arnesen T, Dominguez R (2020) Mechanism of actin N-terminal acetylation. Sci Adv 6(15):eaay8793
Meyer-Schwesinger C (2019) The ubiquitin–proteasome system in kidney physiology and disease. Nat Rev Nephrol 15(7):393–411
Mitta M, Ohnogi H, Yamamoto A, Kato I, Sakiyama F, Tsunasawa S (1992) The primary structure of porcine aminoacylase 1 deduced from cDNA sequence. J Biochem 112(6):737–42
Lin X, Wei M, Song F, Xue D, Wang Y (2020) N-acetylcysteine (NAC) attenuating apoptosis and autophagy in RAW264. 7 cells in response to incubation with mycolic acid from bovine Mycobacterium tuberculosis complex. Pol J Microbiol 69(2):223
Matsumoto S, Häberle J, Kido J, Mitsubuchi H, Endo F, Nakamura K (2019) Urea cycle disorders—update. J Hum Genet 64(9):833–847
Velinov M, Zellers N, Styles J, Wisniewski K (2007) Homozygosity for mutation G212A of the gene for aspartoacylase is associated with atypical form of Canavan’s disease. Clin Genet 73(3):288–289
Sommer A, Christensen E, Schwenger S, Seul R, Haas D, Olbrich H, Omran H, Sass JO (2011) The molecular basis of aminoacylase 1 deficiency. Biochim Biophys Acta Mol Basis Dis BBA-MOL BASIS DIS 1812(6):685–690
Michelucci R, Mecarelli O, Bovo G, Bisulli F, Testoni S, Striano P, Striano S, Tinuper P, Nobile C (2007) A de novo LGI1 mutation causing idiopathic partial epilepsy with telephone-induced seizures. Neurology 68(24):2150–2151
Acknowledgements
We thank the families’ patients to participate in this study. The authors are especially thankful to the personnel of the DeNA laboratory (https://dna-lab.ir/) and Pardis Gene Technology company (http://www.pardisgene.com) for supporting the study. The authors appreciate the contribution of the staffs of Medical Genetics Department of Medical Sciences Faculty, Tarbiat Modares University, Tehran, Iran.
Funding
The results of this publication were supported and funded by Tehran University of Medical Sciences (grant number: 96-02-30-35551). In addition, the National Institute for Medical Research Development (Proposal No. 983886) provided financial and logistic support for this study but had no role in study design, data collection and analysis, data interpretation, and writing of the report, or in making decision to submit the article for publication.
Author information
Authors and Affiliations
Contributions
MG, ART, and PM conceived and designed the experiments. MFM, AD, KZ, SMK, SS, MRA, MH, and AHB conducted the experiments. MG, MFM, AD, ART, and SMK analyzed and interpreted the data. ART, MG, and MFM contributed reagents/materials/analysis tools. AHB, AD, KZ, SMK, SS, and MFM wrote the paper. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Ethics approval
Ethics approval for the study protocol was confirmed by The Human Ethics Committee of Tarbiat Modares University.
Inform consent
Written consent was obtained from parents as legal guardians of the proband.
Consent for publication
The study was performed following the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Mohammad Farid Mohammadi and Ali Dehghani contributed equally to this work.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Mohammadi, M.F., Dehghani, A., Zarabadi, K. et al. Persistent basal ganglia involvement in aminoacylase-1 deficiency: expanding imaging findings and review of literature. Ir J Med Sci 193, 449–456 (2024). https://doi.org/10.1007/s11845-023-03452-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11845-023-03452-0