
Abstract
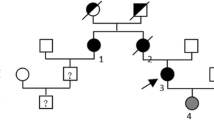
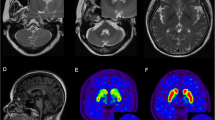
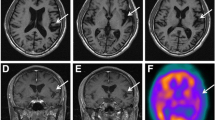
Alexander disease (AxD) is a rare autosomal dominant leukodystrophy caused by heterozygous mutations in the glial fibrillary acid protein (GFAP) gene. The age of symptoms onset ranges from infancy to adulthood, with variable clinical and radiological manifestations. Adult-onset AxD manifests as a chronic and progressive condition, characterized by bulbar, motor, cerebellar, and other clinical signs and symptoms. Neuroradiological findings typically involve the brainstem and cervical spinal cord. Adult-onset AxD has been described in diverse populations but is rare in Israel. We present a series of patients diagnosed with adult-onset AxD from three families, all of Jewish Syrian descent. Five patients (4 females) were diagnosed with adult-onset AxD due to the heterozygous mutation c.219G > A, p.Met73Ile in GFAP. Age at symptoms onset ranged from 48 to 61 years. Clinical characteristics were typical and involved progressive bulbar and gait disturbance, followed by pyramidal and cerebellar impairment, dysautonomia, and cognitive decline. Imaging findings included medullary and cervical spinal atrophy and mostly infratentorial white matter hyperintensities. A newly recognized cluster of adult-onset AxD in Jews of Syrian origin is presented. This disorder should be considered in differential diagnosis in appropriate circumstances. Genetic counselling for family members is required in order to discuss options for future family planning.


Similar content being viewed by others
Data availability
The data used to support the findings of this study are available from the corresponding author upon request.
References
Brenner M, Johnson AB, Boespflug-Tanguy O et al (2001) Mutations in GFAP, encoding glial fibrillary acidic protein, are associated with Alexander disease. Nat Genet 27:117–120. https://doi.org/10.1038/83679
Russo LS, Aron A, Anderson PJ (1976) Alexander’s disease: a report and reappraisal. Neurology 26:607–614. https://doi.org/10.1212/wnl.26.7.607
Pridmore CL, Baraitser M, Harding B et al (1993) Alexander’s disease: clues to diagnosis. J Child Neurol 8:134–144. https://doi.org/10.1177/088307389300800205
Springer S, Erlewein R, Naegele T et al (2000) Alexander disease–classification revisited and isolation of a neonatal form. Neuropediatrics 31:86–92. https://doi.org/10.1055/s-2000-7479
Prust M, Wang J, Morizono H et al (2011) GFAP mutations, age at onset, and clinical subtypes in Alexander disease. Neurology 77:1287–1294. https://doi.org/10.1212/WNL.0b013e3182309f72
Srivastava S, Waldman A, Naidu S (1993) Alexander Disease. In: Adam MP, Everman DB, Mirzaa GM et al (eds) GeneReviews®. University of Washington, Seattle, Seattle (WA)
Barkovich AJ, Messing A (2006) Alexander disease: not just a leukodystrophy anymore. Neurology 66:468–469. https://doi.org/10.1212/01.wnl.0000200905.43191.4d
Caroli F, Biancheri R, Seri M et al (2007) GFAP mutations and polymorphisms in 13 unrelated Italian patients affected by Alexander disease. Clin Genet 72:427–433. https://doi.org/10.1111/j.1399-0004.2007.00869.x
Graff-Radford J, Schwartz K, Gavrilova RH et al (2014) Neuroimaging and clinical features in type II (late-onset) Alexander disease. Neurology 82:49–56. https://doi.org/10.1212/01.wnl.0000438230.33223.bc
Farina L, Pareyson D, Minati L et al (2008) Can MR imaging diagnose adult-onset Alexander disease? AJNR Am J Neuroradiol 29:1190–1196. https://doi.org/10.3174/ajnr.A1060
Messing A (2018) Alexander disease. In: Handbook of Clinical Neurology. Elsevier, pp 693–700. https://doi.org/10.1016/B978-0-444-64076-5.00044-2
Messing A, Brenner M, Feany MB et al (2012) Alexander disease. J Neurosci 32:5017–5023. https://doi.org/10.1523/JNEUROSCI.5384-11.2012
Yoshida T, Sasaki M, Yoshida M et al (2011) Nationwide survey of Alexander disease in Japan and proposed new guidelines for diagnosis. J Neurol 258:1998–2008. https://doi.org/10.1007/s00415-011-6056-3
Nishri D, Edvardson S, Lev D et al (2014) Diagnosis by whole exome sequencing of atypical infantile onset Alexander disease masquerading as a mitochondrial disorder. Eur J Paediatr Neurol 18:495–501. https://doi.org/10.1016/j.ejpn.2014.03.009
Ciammola A, Sangalli D, Sassone J et al (2019) A novel mutation of GFAP causing adult-onset Alexander disease. Front Neurol 10:1124. https://doi.org/10.3389/fneur.2019.01124
Hayashi Y, Nagasawa M, Asano T et al (2017) Central hypothermia associated with Alexander disease. A case report. Clin Neurol Neurosurg 157:31–33. https://doi.org/10.1016/j.clineuro.2017.03.013
Sreedharan J, Shaw CE, Jarosz J, Samuel M (2007) Alexander disease with hypothermia, microcoria, and psychiatric and endocrine disturbances. Neurology 68:1322–1323. https://doi.org/10.1212/01.wnl.0000259543.95222.9d
Kim B, Kim S, Jin MS (2018) Crystal structure of the human glial fibrillary acidic protein 1B domain. Biochem Biophys Res Commun 503:2899–2905. https://doi.org/10.1016/j.bbrc.2018.08.066
Flint D, Li R, Webster LS et al (2012) Splice site, frameshift, and chimeric GFAP mutations in Alexander disease. Hum Mutat 33:1141–1148. https://doi.org/10.1002/humu.22094
Hsiao VC, Tian R, Long H et al (2005) Alexander-disease mutation of GFAP causes filament disorganization and decreased solubility of GFAP. J Cell Sci 118:2057–2065. https://doi.org/10.1242/jcs.02339
Der Perng M, Su M, Wen SF et al (2006) The Alexander disease-causing glial fibrillary acidic protein mutant, R416W, accumulates into Rosenthal fibers by a pathway that involves filament aggregation and the association of alpha B-crystallin and HSP27. Am J Hum Genet 79:197–213. https://doi.org/10.1086/504411
Gorospe JR, Naidu S, Johnson AB et al (2002) Molecular findings in symptomatic and pre-symptomatic Alexander disease patients. Neurology 58:1494–1500. https://doi.org/10.1212/wnl.58.10.1494
Li R, Johnson AB, Salomons G et al (2005) Glial fibrillary acidic protein mutations in infantile, juvenile, and adult forms of Alexander disease. Ann Neurol 57:310–326. https://doi.org/10.1002/ana.20406
Balbi P, Salvini S, Fundarò C, et al (2010) The clinical spectrum of late-onset Alexander disease: a systematic literature review. J Neurol 257:1955–1962. https://doi.org/10.1007/s00415-010-5706-1
Brenner M (2014) Alexander’s Disease. In: Aminoff MJ, Daroff RB (eds) Encyclopedia of the Neurological Sciences, 2nd Edn. Academic Press, Oxford, pp 106–109
Stumpf E, Masson H, Duquette A et al (2003) Adult Alexander disease with autosomal dominant transmission: a distinct entity caused by mutation in the glial fibrillary acid protein gene. Arch Neurol 60:1307–1312. https://doi.org/10.1001/archneur.60.9.1307
Yoshida T, Mizuta I, Saito K et al (2013) Effects of a polymorphism in the GFAP promoter on the age of onset and ambulatory disability in late-onset Alexander disease. J Hum Genet 58:635–638. https://doi.org/10.1038/jhg.2013.83
Hagemann TL, Powers B, Mazur C, et al (2018) Antisense suppression of glial fibrillary acidic protein as a treatment for Alexander disease: GFAP ASO Therapy in AxD. Ann Neurol 83:27–39. https://doi.org/10.1002/ana.25118
Hagemann TL, Powers B, Lin N-H et al (2021) Antisense therapy in a rat model of Alexander disease reverses GFAP pathology, white matter deficits, and motor impairment. Sci Transl Med 13:eabg4711. https://doi.org/10.1126/scitranslmed.abg4711
Author information
Authors and Affiliations
Contributions
SA, SL, LG, and SHB played a key role in the conception and design of the study. SA, TFK, FS, and SHB monitored patients and provided valuable clinical insights. NM, OB, and DD conducted the genetic testing and bioinformatic analysis. OLS contributed to the interpretation of the neuroradiological data. OC, EP, and LG provided genetic expertise for the study. SA, LG, and SHB drafted the manuscript. All the authors critically reviewed the manuscript, provided input, and approved the final version.
Corresponding author
Ethics declarations
Consent to participate
Informed consent was obtained from all individual participants included in the study, and if deceased—from their legal guardian.
Competing interests
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Anis, S., Fay-Karmon, T., Lassman, S. et al. Adult-onset Alexander disease among patients of Jewish Syrian descent. Neurogenetics 24, 303–310 (2023). https://doi.org/10.1007/s10048-023-00732-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10048-023-00732-w