
Incretin Mimetics Restore the ER-Mitochondrial Axis and Switch Cell Fate Towards Survival in LUHMES Dopaminergic-Like Neurons: Implications for Novel Therapeutic Strategies in Parkinson’s Disease
Abstract
Background:
Parkinson’s disease (PD) is a progressive neurodegenerative movement disorder that afflicts more than 10 million people worldwide. Available therapeutic interventions do not stop disease progression. The etiopathogenesis of PD includes unbalanced calcium dynamics and chronic dysfunction of the axis of the endoplasmic reticulum (ER) and mitochondria that all can gradually favor protein aggregation and dopaminergic degeneration.
Objective:
In Lund Human Mesencephalic (LUHMES) dopaminergic-like neurons, we tested novel incretin mimetics under conditions of persistent, calcium-dependent ER stress.
Methods:
We assessed the pharmacological effects of Liraglutide—a glucagon-like peptide-1 (GLP-1) analog—and the dual incretin GLP-1/GIP agonist DA3-CH in the unfolded protein response (UPR), cell bioenergetics, mitochondrial biogenesis, macroautophagy, and intracellular signaling for cell fate in terminally differentiated LUHMES cells. Cells were co-stressed with the sarcoplasmic reticulum calcium ATPase (SERCA) inhibitor, thapsigargin.
Results:
We report that Liraglutide and DA3-CH analogs rescue the arrested oxidative phosphorylation and glycolysis. They mitigate the suppressed mitochondrial biogenesis and hyper-polarization of the mitochondrial membrane, all to re-establish normalcy of mitochondrial function under conditions of chronic ER stress. These effects correlate with a resolution of the UPR and the deficiency of components for autophagosome formation to ultimately halt the excessive synaptic and neuronal death. Notably, the dual incretin displayed a superior anti-apoptotic effect, when compared to Liraglutide.
Conclusions:
The results confirm the protective effects of incretin signaling in ER and mitochondrial stress for neuronal degeneration management and further explain the incretin-derived effects observed in PD patients.
INTRODUCTION
Parkinson’s disease (PD) is a heterogenous neurogenerative movement disorder, involving the progressive manifestation of resting tremor, rigidity, bradykinesia, and postural instability, as well as non-motor, cognitive impairments. At a tissue level, PD features the gradual degeneration of dopaminergic neurons in the substantia nigra pars compacta along with the accumulation of aggregated forms (oligomers and filaments) of α-synuclein in neurites (known as Lewy neurites) and in neuron soma (known as Lewy bodies). Although the PD etiology and pathogenic development remain elusive, varying degrees of the interplay between genomic predispositions, environmental factors, and aging seem to underlie dopaminergic degeneration [1] and all converge into a dysfunctional proteostasis network, perturbed mitochondrial function, and dysregulated calcium fluxes [2–4].
The buffering capacity and integrity of all these processes lie in the homeostasis of the endoplasmic reticulum (ER) [5–7]. The ER is a dynamic membrane system that extends from the soma to the entire dendrite arbor, including some dendritic spines, and the axon of the neuron [8] that, in a dynamic cross-talk with most intercellular organelles via membrane contact sites, provides the major platform for autophagosome [9, 10] and mitochondrial biogenesis [11, 12]. The ER is additionally the fundamental sub-cellular compartment for protein synthesis and quality control of approximately one-third of the total proteome, including all plasma membrane channels and receptors important for synaptic function [6, 13]. It also is the major calcium storage depot in the cell [14]. Calcium release from the ER mediates neurotransmitter exocytosis at the pre-synapse whilst potentiating gene transcription and modulating membrane excitability and dendritic spine structure for post-synaptic plasticity [14–16]. Upon release, the sarcoplasmic/ER calcium ATPase (SERCA) channel buffers calcium reuptake from the cytosol into the ER to replenish the local calcium stores [14].
As such, a potential depletion of ER calcium stores will preclude the functionality of local molecular chaperones that closely monitor and assist folding, maturation, and the delivery of secretory and membrane proteins. The former results in a rise of the unfolded or misfolded protein load within the organelle lumen, a cellular state defined as ER stress [13]. In turn, the cell activates an adaptive signaling network, known as the unfolded protein response (UPR). The prototypic UPR engages the three ER-resident transmembrane stress transducers—the protein kinase RNA-like ER kinase (PERK), activating transcription factor 6 (ATF6), and inositol-requiring enzyme 1 (Ire1), which initially attenuate global protein synthesis, potentiate the transcription of chaperone proteins, and degrade the abnormal protein load through the proteasome (ER-associated degradation) and lysosome-mediated autophagy to safeguard proteostasis [5, 13]. ER stress further stimulates an early increase in mitochondrial respiration that depends crucially upon intraorganellar coupling and calcium transfer and establishes the bioenergetic sources required for the adaptation to this response [17]. However, upon stress chronicity, the UPR adapts its dynamics and shifts cell fate towards apoptosis through diverse but often overlapping mechanisms, including autophagy fail, bioenergetic crisis, the transcription of CAAT/enhancer-binding protein (C/EBP) homologous protein (Chop), caspase activation, and aberrant expression and activity patterns of Bcl-2 family members along with their mediators [5, 13].
It is therefore intuitive that therapeutic interventions which mitigate the UPR towards a balance between protein generation and degradation and promote homeostasis along the ER/mitochondrial axis may benefit the clinical outcome of neurodegenerative disorders [5, 6]. With this regard, the present study addresses the restorative effects of the GLP-1 analog Liraglutide and the dual GLP-1/GIP incretin analog DA–CH3 in dopaminergic-like neurons. Glucagon-like peptide (GLP-1) is a hormone and growth factor that has been shown to exert neuroprotective effects in preclinical and clinical studies [18–20]. Exenatide is a GLP-1 mimetic that has shown good neuroprotective effects in patients with PD, and a phase 3 trial is currently ongoing [21, 22]. A phase 2 clinical trial testing the GLP-1 analogue Liraglutide has shown similar protective effects [23], and further trials testing other GLP-1 mimetics are ongoing [18]. In addition, a phase 2 clinical trial testing Liraglutide in Alzheimer’s disease (AD) patients showed improvements in memory and cognition [24]. These first clinical results demonstrate that this novel approach is effective and viable, but more information on the underlying mechanisms of action is needed. GIP is glucose-dependent insulinotropic polypeptide, the sister incretin hormone of GLP-1 that previously showed good neuroprotective effects on its own [25].
More specifically, we employed the human embryonic neuronal precursor LUHMES cells to obtain post-mitotic dopaminergic-like neurons and assessed the pharmacological effects of Liraglutide and dual incretin analog DA—CH3 in a) mitochondrial function and cell bioenergetics, b) UPR and autophagy, c) synaptic damage, and d) cytotoxicity under conditions of persistent ER stress. Herein, we provoked ER stress and UPR by lowering the ER calcium levels by the irreversible SERCA inhibitor, thapsigargin. The stressor was chosen not only based on the well-documented role of calcium dynamics in neuronal physiology and disease [4, 14]. Most notably, it was selected, considering the fundamental role of the SERCA channel in maintaining the required steep calcium gradients within the different neuronal intracellular compartments (resting cytosolic calcium concentration: nanomolar range vs. calcium concertation in the ER: millimolar range) and its pathognomonic implication in the α-synuclein aggregation [4, 26]. Although, we have previously established that the activation of GLP-1 receptor can resolve UPR and mitigate the downstream apoptotic signals in the human neuroblastoma SH-SY5Y cells [27], the proliferating and cancerous nature of these cells pose concerns on the data extrapolation in post-mitotic neurons. Furthermore, we have previously reported the neurorestorative effects of the dual incretin DA—CHE in aberrant cognitive functioning and proteostasis of the APPSWE/PS1ΔE9 mouse model of AD [28]. Nevertheless, intraspecies difference should be considered in the data translation to clinic. Therefore, this study comes to fill all these gaps and deepen our understanding of incretin-driven neuroprotection in human disease.
MATERIALS AND METHODS
Materials
The Cell proliferation kit II (2,3-Bis-(2-methoxy-4-nitro-5-sulphophenyl)-2H-tetrazolium-5-carboxanilide (XTT)) and Cytotoxicity Detection KitPLUS (lactate dehydrogenase (LDH)) were purchased from Roche Diagnostics Ltd (West Sussex, UK). Agilent Seahorse XF Glycolytic Rate Assay (103344-100) and Cell Mito Stress Test (103015-100) kits, Seahorse XF24 cell culture microplates, and the corresponding Seahorse XF assay media and calibrant solution were acquired from Agilent Technologies AG (Basel-Stadt, Switzerland). Bovine serum albumin (BSA), tris buffered saline (TBS; pH 8.0) supplemented or not with 0.05% Tween 2®, phosphate buffered saline (PBS; pH 7.4), fibronectin from human plasma (0.1%), poly-L-ornithine solution (MW = 30,000–70,000; 0.1 mg mL–1), N6 2’-O-Dibutyryl cAMP (adenosine-3’,5’-cyclic monophosphate) sodium salt, tetracycline hydrochloride (BioReagent, suitable for cell culture), dimethyl sulfoxide (DMSO; anhydrous, ≥99.9%), and para-formaldehyde were obtained from Sigma-Aldrich Corporation (Dorset, UK). Human recombinant glial-derived neurotrophic factor (GDNF) was obtained from R&D Systems (Bio-Techne Ltd, Oxfordshire, UK). Quick Start™ Bradford protein assay kit was obtained from BIO-RAD Laboratories Ltd (Hertfordshire, UK). MitoBiogenesis™ In-Cell ELISA Kit (Colorimetric; ab110217), JC-1 – Mitochondrial Membrane Potential Assay Kit (ab113850), and normal goat serum were purchased from Abcam (Cambridgeshire, UK). VECTASHIELD antifade mounting medium with DAPI were purchased from Vector Laboratories Ltd (Cambridgeshire, UK). Other materials and reagents for cell culture, western blotting, and immunocytochemistry were obtained from Fisher Scientific UK Ltd (Leicestershire, UK), unless otherwise stated.
Cell culture
The Lund human mesencephalic (LUHMES) cell line is a sub-clone of the tetracycline-controlled, v-myc-over-expressing fetal human mesencephalic-derived cell line MESC2.10, established and characterized at Lund University (Lund, Sweden) [18, 19]. The LUHMES cell line (ATCC® CRL-2927™, RRID: CVCL_B056) was obtained from LGC Standards. Proliferating LUHMES cells were cultured in Advanced DMEM / F-12 supplemented with 1X Glutamax™, 1X N-2 supplement, and 40 ng mL–1 basic recombinant human fibroblast growth factor (bFGF) (hereinafter referred to as complete growth medium) in culture vessels, which were sequentially pre-coated with 50μg mL–1 poly-L-ornithine and 1 g mL–1 fibronectin to promote cell attachment. Cells were maintained at 37°C in a humidified incubator with 5% CO2 and 95% air. Culture medium was renewed every 1 to 2 days.
Coating procedure for culture vessels
Nunc™ Cell Culture Treated EasY Flasks, Nunc™ MicroWell™ flat-bottomed 96-well plates, Seahorse XF24 cell culture microplates, and Millicell EZ SLIDE eight-well glass chamber slides were pre-coated overnight with 50μg mL–1 poly-L-ornithine at room temperature. Following the removal of the poly-L-ornithine solution, the culture vessels were rinsed three times with Gibco® Water for Injection for Cell Culture and allowed to air-dry uncapped in a biological safety cabinet. Subsequently, the culture vessels were coated with 1 g mL–1 fibronectin for 3 h at 37°C. Fibronectin solution was then removed, and all the vessels were rinsed three times with Gibco® Water for Injection for Cell Culture and allowed to air-dry uncapped in a biological safety cabinet. When dry, the culture vessels were filled with the appropriate culture medium and placed for at least 30 min in the incubator to allow the medium to reach its normal pH (7.0–7.4) and avoid excessive alkalinity of medium during cell handling procedures. Alternatively, they were sealed and stored under aseptic conditions in 4°C for up to a week. Poly-L-ornithine and fibronectin were diluted at the required concentration in Gibco® Water for Injection for Cell Culture and Hank’s balanced salt solution (HBSS), respectively, immediately before used.
Sub-culturing procedure
Proliferating LUHMES cells were sub-cultured when 70–80% confluent and grown up to passage 10 to avoid senescence and loss of pluripotency. Initially, the spent culture medium was discarded and the cell monolayer was gently rinsed with Dulbecco’s phosphate-buffered saline (DPBS) that was formulated without calcium and magnesium. Subsequently, the cell monolayer was incubated with the TrypLE™ Select CTS™ dissociation reagent at 37°C for ∼5 min. Cells were collected by centrifugation at 200× g for 7 min and suspended in complete growth medium for the cell line. Viable cells were counted and seeded at the desired cell density for the differentiation procedure using the Countess™ Automated Cell Counter. Alternatively, proliferating LUHMES cells were seeded at 1:10 ratio.
Differentiation procedure
Differentiation procedure was conducted as previously described [20]. Briefly, 1×107 proliferating LUHMES cells were seeded into pre-coated T175 Nunc™ Cell Culture Treated EasY Flasks in the appropriate complete growth medium. Twenty-four hours post seeding, spent medium was discarded and replaced with the differentiation medium that defines day 0 (d0) of the differentiation procedure. Differentiation medium consisted of Advanced DMEM/F12 medium supplemented with 1X Glutamax™, 1X N-2 supplement, 2 ng mL–1 human recombinant GDNF, 1 mM N6 2’-O-Dibutyryl cAMP sodium salt, and 1μg mL–1 tetracycline hydrochloride. At d2, pre-differentiated LUHMES cells were sub-cultured at the density of 2×105 cells ml–1 into the corresponding culture vessels for each assay. Differentiation process continued for additional 3 to 4 days to allow cells to morphologically and biochemically mature into post-mitotic neurons. Differentiation medium was changed at d4 of the differentiation procedure.
Cell treatments
All agents for cell treatment were handled as previously described [27, 28] and introduced to neurons at d 6 of the differentiation, at least in triplicate per assay per experiment.
Thapsigargin is a naturally occurring sesquiterpene lactone that selectively inhibits SERCA, triggering a transient increase in the cytosolic calcium and depleting ER calcium stores [29]. It was reconstituted in 100% DMSO at a stock concentration of 1 mM, aliquoted and stored at –20°C until used. For the experiments, thapsigargin stock preparations were serially diluted in differentiation medium at final working concentrations of 100 nM, containing 0.01% DMSO; DMSO concentration up to 0.33% has no impact on LUHMES cell viability and neurite outgrowth [24]. Neurons were incubated with thapsigargin for 16 h for all the assays. The scheme of thapsigargin cell treatment (concentration and incubation) was chosen based on our previously published data in human neuroblastoma SH-SY5Y cell line [27].
The incretin mimetics were purchased from China Peptides Co., Ltd and Biochem Ltd (Shanghai, China). The purity of the peptides was analyzed by reverse-phase high performance liquid chromatography (HPLC) and characterized using matrix-assisted laser desorption/ionization time-of-flight (MALDI–TOF) mass spectrometry. The purity of the peptides tested was ≥96% in accord with the manufacturers’ reports. The amino acid sequences of the peptides tested herein are described in Table 1. The peptides were maintained in powdered, desiccated form at –80°C and reconstituted in Gibco® Water for Injection for Cell Culture to a concentration of 1 mg ml–1, aliquoted and stored at –20°C until used. For the experiments, Liraglutide and GLP-1/GIP Dual Agonist stock preparations were diluted in differentiation medium to a final working concentration of 100 nM. The concentration was selected on the basis of previous experiments in which our group has established optimal working concentrations for the neuroprotective and anti-apoptotic effects of incretin mimetics [27].
Table 1
Amino acid sequence and molecular weight (MW) of the incretin mimetics used in the current study
| Name | Amino acid sequence | MW (Da) | Reference |
| Liraglutide | HAEGTFTSDVSSYLEGQAA [Lys-γE-C16 acyl] EFIAWLVRGRG–OH | 3751.26 | [22] |
| GLP-1/GIP Dual Agonist | YXEGTFTSDYSIYLDKQAAXEFV NWLLAGGPSSGAPPPS–NH2 | 4234.63 | [23] |
Wherein the sequences, the “X” symbol stands for aminoisobutyric acid.
Assessment of neuronal cell viability
Sixteen hours post-treatment initiation, we quantified neuronal cell viability with the Cell proliferation kit II (XTT) and Cytotoxicity Detection KitPLUS (LDH), as per our previously published methodology [21, 25, 26].
Assessment of neuronal bioenergetics
Extracellular flux (XF) analysis was employed for the real-time quantification of mitochondrial respiration and glycolysis in live neurons. Specifically, we determined the oxygen consumption rate (OCR) owing to ATP turnover, proton leak and maximal respiratory capacity and the proton efflux rate (PER) from glycolytic and mitochondrial-derived acidification with the Agilent Seahorse XF Cell Mito Stress Test kit and Agilent Seahorse XF Glycolytic Rate Assay kit, respectively. Both assays were formatted into pre-coated Seahorse XF24 cell-culture microplates and performed as previously described [30, 31].
Assessment of mitochondrial membrane potential (ΔΨm)
We assessed ΔΨm with the 5,5’,6,6’-Tetrachloro-1,1’,3,3’-tetraethyl-benzimidazolylcarbocyanine iodide (JC-1). JC-1 is a lipophilic, cationic dye that naturally exists in monomers and fluoresces in the green spectrum. JC-1 typically enters and accumulates into the energized and negatively-charged mitochondria and there, in a concentration-dependent manner, forms reversible complexes (aggregates) that emit signal at the red spectrum. However, under unhealthy conditions during which mitochondria feature an increased permeability and the loss of electrochemical potential, JC-1 does not reach the sufficient concentration for the formation of aggregates and thus retains its original green-fluorescent monomeric form. The ratio of the red-to-green fluorescence signal that reflects the ratio of the aggregated over the monomeric forms of JC-1 is an indicator of mitochondria polarization[32].
The assay was formatted into pre-coated Nunc™ MicroWell™ flat-bottomed black 96-well plates. Sixteen hours post-treatment initiation, neurons were washed once with pre-warmed 1X dilution buffer (kit provided) and incubated with 20μM JC-1 for 10 min at 37°C in a humidified incubator with 5% CO2 and 95% air. Following JC-1 staining and two subsequent washes with pre-warmed 1X dilution buffer, microplates were read with Infinite® 200 PRO microplate reader at λexcitation= 535 nm and λemission= 590 nm and at λexcitation= 475 nm and λemission= 530 nm to monitor aggregated and monomer forms of the dye, respectively. Acquired fluorescent signal for each JC-1 form was initially normalized to the background and then used for calculating the ratio of aggregated to monomer JC-1. The latter was subsequently normalized to the corresponding (per condition) Janus Green cell normalization stain. Janus green dye (ab111622) was obtained by Abcam (Cambridgeshire, UK) and the staining was performed in accord to the manufacturer’s protocol.
Assessment of mitochondrial biogenesis
In-cell colorimetric ELISA assay was employed for quantitative measurement of the protein expression ratio of the cytochrome c oxidase subunit I (COX1; mitochondrial DNA-encoded protein) to the succinate dehydrogenase (SDH-A; nuclear DNA-encoded protein) to monitor mitochondrial biogenesis. The assay was formatted into pre-coated Nunc™ MicroWell™ flat-bottomed transparent 96-well plates. Sixteen hours post-treatment initiation, neurons were fixed in 4% paraformaldehyde in 1X PBS for 10 min at room temperature and subsequently washed thrice with 1X PBS. Endogenous alkaline phosphatase activity was blocked with a 5 min-incubation with 0.5% acetic acid and neurons were then permeabilized in 1% Triton-X-100 in 1X PBS for 30 min at room temperature with gentle agitation. After a two-hour blocking step, neurons were probed with the provided primary antibody cocktail overnight at 4°C with gentle agitation. The following day, neurons were briefly washed with PBS supplemented with 2% Tween®-20 and incubated with the provided AP/HRP-labelled secondary antibody solution for 1 h at room temperature with gentle agitation. AP and HRP development were respectively used for the kinetic detection of the protein levels of SDH-A and COX1 at 405 and 600 nm over a cycle of 15 min with an inter-measurement interval timeof ∼60 s.
Immunocytochemistry
Immunocytochemical experiments were formatted into pre-coated Millicell EZ SLIDE eight- well glass chamber slides. Sixteen hours post-treatment initiation, neurons were fixed, permeabilized and stained for Nrf2 in accord to the previously described protocol [27].
Protein extraction and immunoassays
Sixteen hours post-treatment initiation, neurons were washed twice with ice-cold 1X PBS and harvested in 1X PathScan® Sandwich ELISA cell lysis buffer supplemented with protease/phosphatase inhibitor cocktail (1X). After two freeze/thaw cycles, the whole-cell lysate was collected and sonicated for 1 min (5 s ON/5 s OFF – 6 cycles). The total protein was extracted by centrifugation at ∼16000× g at 4°C for 15 min. The Quick start™ Bradford protein assay was conducted to estimate the protein concentration of the samples. Protein samples of whole-cell lysate were aliquoted and stored at –80°C till processed for western blotting and PathScan® Intracellular Signaling ELISA Array (#14471, Cell Signaling Technology, Greater London, UK). Both immunoassays were conducted in accord to our previously published methodology [27]. The primary antibodies used in the western blotting experiments are summarized in Supplementary Table 1. The intercellular signaling targets of the array are listed in Supplementary Figure 1.
Statistics
All the results were expressed as mean±standard error (SEM) of five independent experiments. Assumption of normality was examined with D’Agostino-Pearson’s K-squared and Shapiro-Wilk tests. Differences among means were considered significant if p≤0.05. Data processed with two-way ANOVA analysis, followed by post-hoc Bonferroni’s multiple-comparison t-tests to identify differences among experimental groups and effects of incretin treatments under control and ER-stressed conditions. We employed repeated-measures three-way ANOVA, followed by post-hoc Bonferroni’s multiple-comparison t-tests, to analyze the effects of each incretin in the mitochondrial biogenesis under control and ER-stressed conditions over time. Statistical calculations were performed in GraphPad Prism 8 (GraphPad Software Inc., San Diego, USA) for Mac OS X software.
Research ethics statement
No ethical approval was required for the present study. All experiments, image acquisition, and cell treatments were performed in a blinded fashion.
RESULTS
The GLP-1/GIP dual agonist maximizes the incretin-derived benefit on attenuating the neuronal UPR upon chronic ER stress
We initially quantified the expression of transducers, mediators of the UPR by western blotting, as shown in Fig. 1. Chronic thapsigargin treatment potentiates a two-fold increase in the ER-stress sensor BiP and almost halves the protein levels of the full-length (90 kDa) UPR transducer ATF6 that may signify its truncation and downstream activation [33]. It additionally precludes the engagement of Ire1α kinase of the UPR, evident by the one-fold decrease in the expression levels of the activating phosphorylation at the serine 724 residue (Ser724) over the total protein levels. Furthermore, neurons stressed with thapsigargin overnight featured a significantly decreased expression of the chaperones calnexin and protein disulfide isomerase (PDI), while the ER oxidoreductase 1α (ERO1-Lα) is upregulated. These findings indicate a perturbation of the oxidative folding machinery for protein quality control within the ER lumen. Also, thapsigargin-treated neurons display a seven-fold rise in the levels of the transcription factor Chop that is accompanied by an upregulation of the active fragment at 42-kDa of caspase12 (CASP12); these biochemical footprints altogether signal for the engagement of the apoptotic phase of the neuronal UPR [2, 3, 5, 13].
Fig. 1
Liraglutide (LIRA) and the GLP-1/GIP dual agonist (DA) both attenuate the neuronal unfolded protein response (UPR) and restore folding capacity within the endoplasmic reticulum (ER) lumen, with the dual activation of the GLP-1 and GIP receptors superiorly restoring the expression of apoptotic UPR mediators. On d 6 of the differentiation period, dopaminergic-like neurons from the human LUHMES cell line were treated with 0 and 100 nM of thapsigargin (TG) in the presence or absence of each incretin tested for 16 h. Neurons were then harvested, and the expression of (i) the UPR transducers – ATF6 (a) and phosphorylated to total Ire1α (b), (ii) the local chaperones BiP (c), calnexin (d), Ero1-Lα (e), and (iii) of the UPR mediators for apoptotic signaling – Chop (g) and caspase 12 (CASP12) (h) were determined by western blotting. β-Actin served as the loading control to our quantitative analyses. Each bar represents the mean±SEM from five independent experiments. Loading controls for b and e, and for c, g, and h are the same. Data are expressed as fold change to the control (CNTRL; unstressed/untreated conditions) and analyzed by two-way ANOVA, followed by post hoc Bonferroni’s multiple comparison t-test: *p≤0.05, **p≤0.01 & ***p≤0.001 compared to CNTRL; ##p≤0.01 & ###p≤0.001 compared to the TG-stressed neurons; ∂∂p≤0.01 compared to the Liraglutide-treated, TG-stressed neurons.
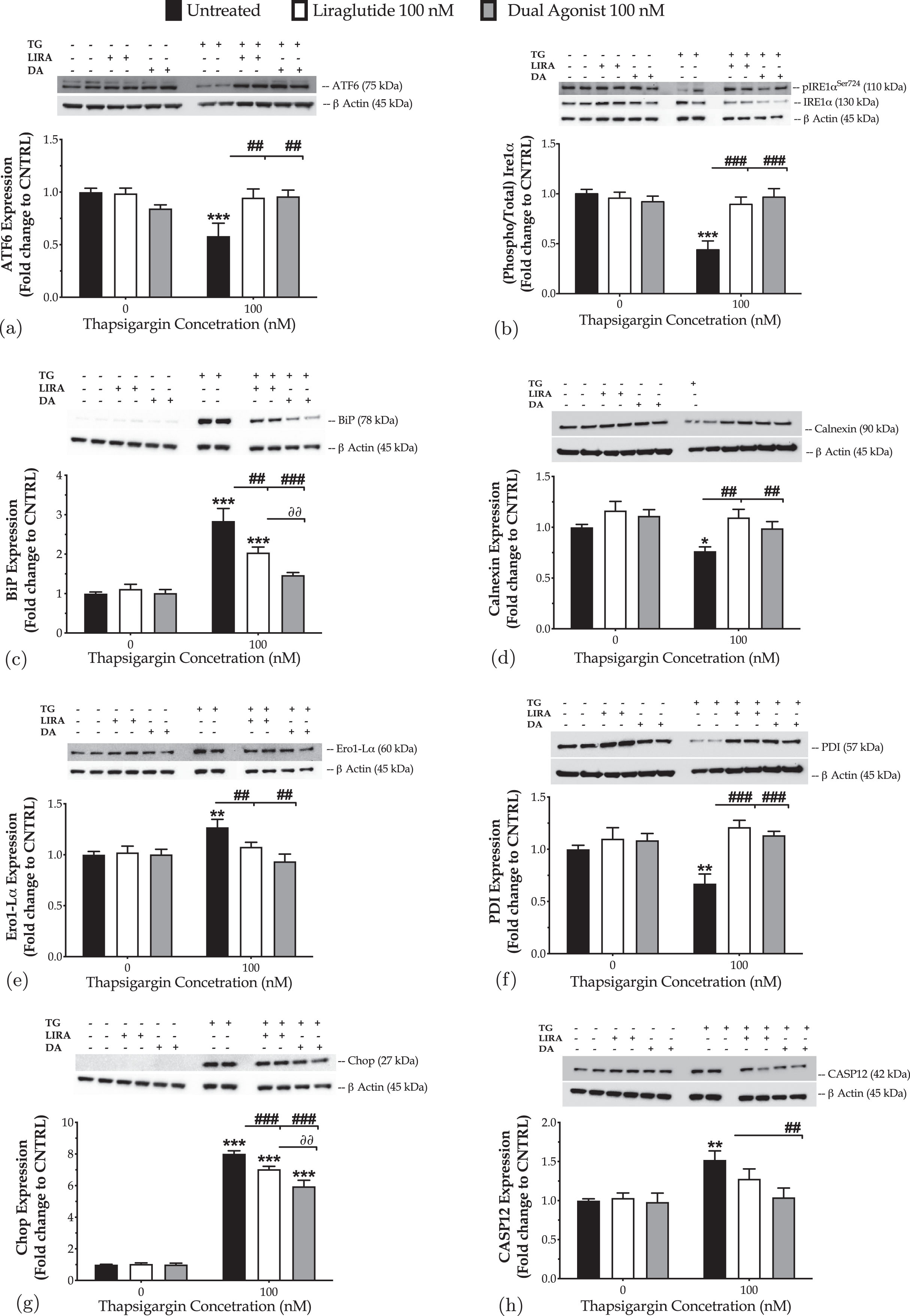
Next, we addressed the pharmacological effects of the GLP-1 in the amelioration/resolution of the ER stress. Two-way ANOVA demonstrates significant interactions between Liraglutide and thapsigargin on the protein levels of ATF6 (F (1,28) = 5.64, p = 0.0247), the phosphorylated at Ser724 over the total Ire1α (F (1,28) = 13.38, p = 0.0010), and BiP (F (1,28) = 6.21, p = 0.0189). Liraglutide co-treatment restores the downregulated expression of full-length ATF6 and activity of Ire1α, as illustrated in Fig. 1a and 1b. It significantly ameliorates the abnormal BiP levels (post-hoc; p≤0.01) of the ER-stressed neurons (Fig. 1c). It further normalizes the aberrant expression of calnexin (Fig. 1d), Ero1-Lα (Fig. 1e), and PDI (Fig. 1f) to favor folding processes within the organelle lumen. These findings are accompanied by a significant drop in the expression levels of Chop (two-way ANOVA interaction: F (1,28) = 14.19, p = 0.0008) (Fig. 1g). However, the expression of the active fragment of CASP12 remains unaffected following Liraglutide co-treatment, though a trend of decrease is prominent in Fig. 1h.
Similarly, the dual agonist reinstates homeostasis for the UPR effectors (two-way ANOVA interaction of dual agonist×thapsigargin: BiP, F (1,28) = 16.80, p = 0.0003; ATF6, F (1,28) = 13.66, p = 0.0009; Ire1α, F (1,28) = 18.12, p = 0.0002) and molecular chaperones for protein quality control (two-way ANOVA interaction of dual agonist×thapsigargin: calnexin, F (1,28) = 10.66, p = 0.0029; Ero1-Lα, F (1,28) = 7.70, p = 0.0097; PDI, F (1,28) = 9.28, p = 0.0050) upon chronic thapsigargin co-treatment (Fig. 1). The dual stimulation of GLP-1 and GIP receptors becomes notably essential for the resolution of the UPR, evident by the BiP normalization (Fig. 1c) in the ER-stressed neurons, when compared to the single GLP-1 activation (two-way ANOVA interaction of the GIP receptor co-stimulation×thapsigargin: F (1,28) = 4.798, p = 0.0370; post hoc, p < 0.01). It is additionally more critical for mitigating UPR apoptotic signals in neurons; the dual incretin produces a more considerable amelioration of the ectopic Chop expression (Fig. 1g) when compared to Liraglutide (two-way ANOVA interaction of GIP receptor co-stimulation×thapsigargin: F (1,28) = 5.732, p = 0.0236). It also brings back to control levels the expression of the cleaved caspase 12(Fig. 1h).
GLP-1/GIP dual agonist balances the aberrant bioenergetics and mitochondrial function to the maximum
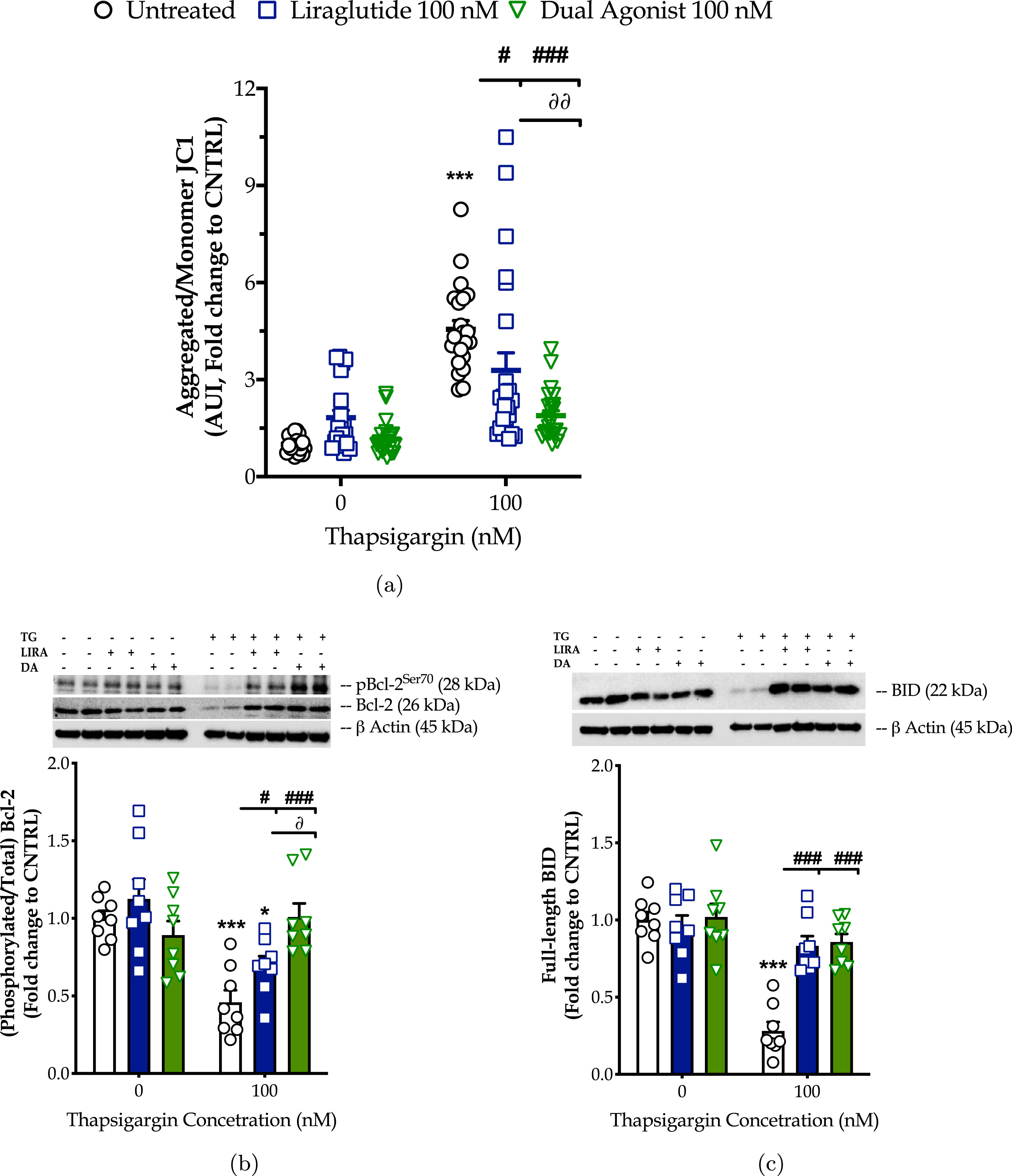
Chronic ER stress with thapsigargin or tunicamycin has been reported to generate a slow, though persistent, calcium flux into the mitochondria that provokes the aberrant membrane permeabilization of the organelle [34]. The latter perturbs the electrochemical gradient for ΔΨm that orchestrates the respiratory rate, ATP synthesis and formation of reactive oxygen species, and thus can potentiate a bioenergetic crisis and mitochondrial dysfunction [35]. As evident in our findings too, persistent ER stress with thapsigargin specifically dissipates the basal and maximal OCR that signals for mitochondrial respiration arrest and renders the neurons incapable and inflexible to meet their energy demands both under baseline and metabolically-challenging conditions (Fig. 2). It decreases by 6-fold the basal and compensatory glycolysis, signifying a shutdown of the anaerobic bioenergetic function, as well (Fig. 3). At the same time, it induces a four-times rise in the ratio of the aggregated over the monomer forms of JC1 (Fig. 4a), indicating an exacerbation of the negative charge within the mitochondrial lumen.
Fig. 2
Liraglutide and the GLP-1/GIP dual agonist restore the arrested oxidative phosphorylation in neurons subjected to persistent ER stress. On d 6 of the differentiation period, dopaminergic-like neurons from the human LUHMES cell line were treated with 0 and 100 nM of thapsigargin (TG) in the presence or absence of each incretin tested for 16 h. We then monitored the oxygen consumption rate (OCR) over the sequential injection of oligomycin (1μM; ATP synthase inhibitor), FCCP (1.5μM; mitochondrial uncoupler) and of Rotenone + Antimycin A (0.5μM; complex I and III inhibitors) with the Seahorse XFe24 Extracellular Flux Analyzer, as illustrated in the representative profile for neuronal mitochondrial respiration (a). Basal, ATP-linked, proton-leak, maximal and non-mitochondrial OCR were subsequently quantified and illustrated as bar graphs (b). Each point and bar represent the mean±SEM from five independent experiments. Data are processed with two-way ANOVA, followed by post hoc Bonferroni’s multiple comparison t-test: *p≤0.05, **p≤0.01 & ***p≤0.001 compared to the control (CNTRL; unstressed/untreated conditions); #p≤0.05 & ##p≤0.01 compared to the TG-stressed neurons.
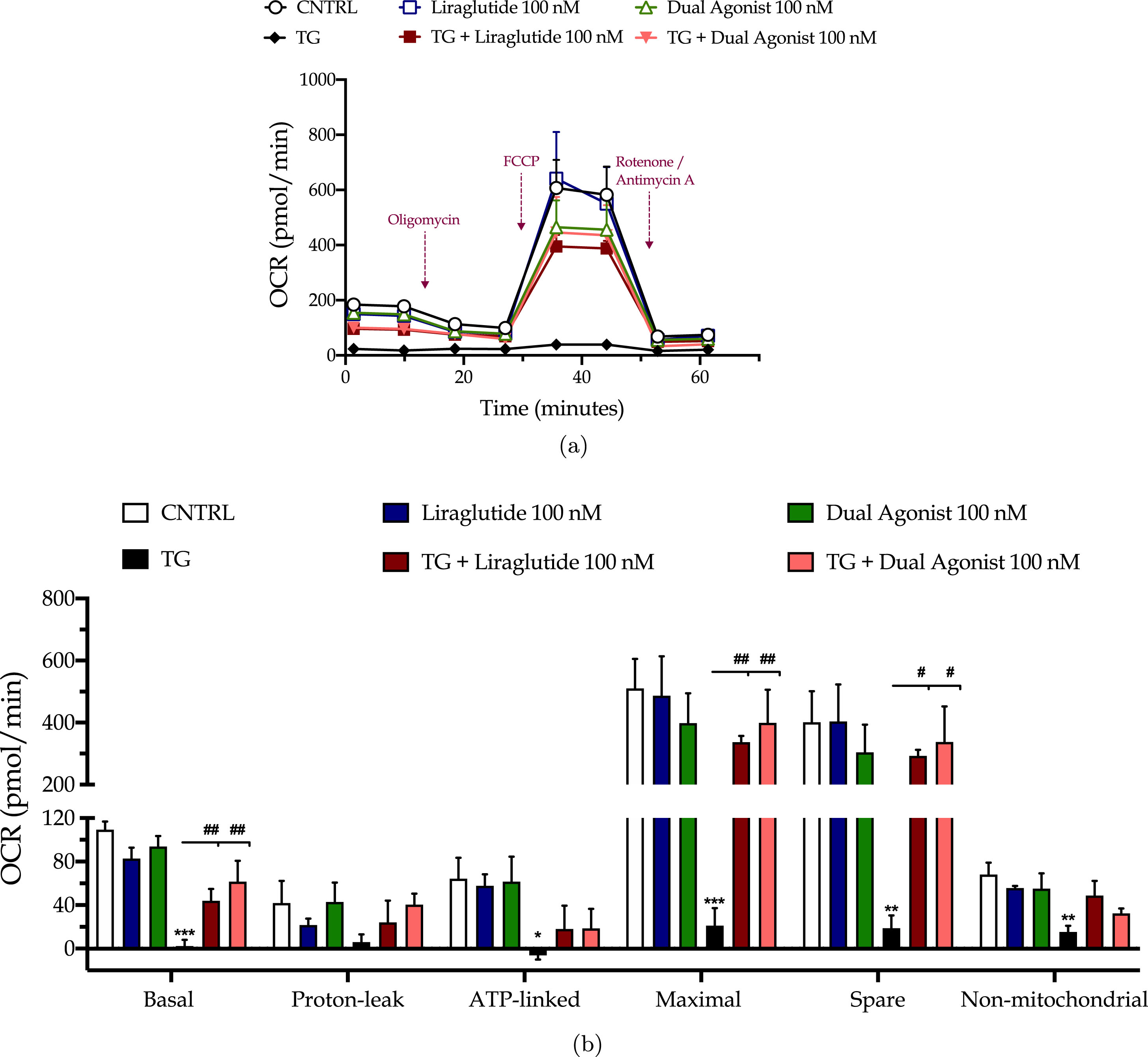
Fig. 3
Incretin co-treatments re-balance the suppressed neuronal glycolysis under conditions of persistent ER stress. On d 6 of the differentiation period, dopaminergic-like neurons from the human LUHMES cell line were treated with 0 and 100 nM of thapsigargin (TG) in the presence or absence of each incretin tested for 16 h. We then quantified the proton efflux rate (PER) over the sequential injection of Rotenone + Antimycin A (to compel the cells to rely on glycolysis for ATP production) and 2-deoxy-D-glucose (20 mM; to inhibit the glycolytic ATP production) with the Seahorse XFe24 Extracellular Flux Analyzer, as illustrated in the representative profile (a). Basal glycolysis, basal PER, and compensatory glycolysis were subsequently calculated and illustrated as bar graphs (b). Each point and bar represent the mean±SEM from five independent experiments. Data are processed with two-way ANOVA, followed by post hoc Bonferroni’s multiple comparison t-test: **p≤0.01 compared to the control (CNTRL; unstressed/untreated conditions); #p≤0.05 compared to the TG-stressed neurons.
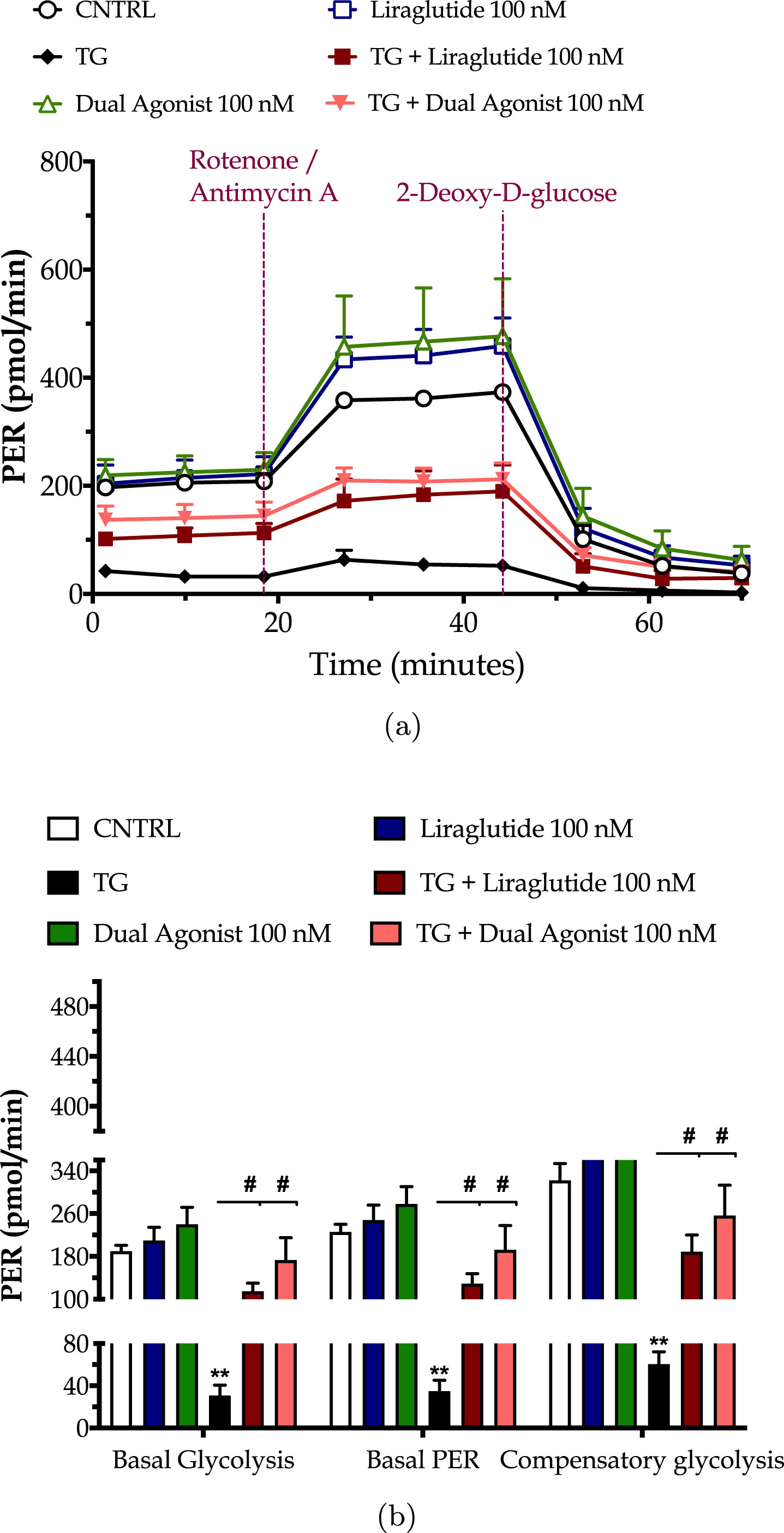
Fig. 4
The GLP-1/GIP dual agonist maximizes the incretin-derived benefit on rescuing the hyperpolarization of the mitochondrial membrane upon chronic neuronal ER stress. On d 6 of the differentiation period, dopaminergic-like neurons from the human LUHMES cell line were treated with 0 and 100 nM of thapsigargin (TG) in the presence or absence of each incretin tested for 16 h. We then probed the neurons with 20μM JC-1 for 10 min and monitored the fluorescence signal of the aggregated over the monomer form of the dye in a microplate reader. Fluorescent signal (in arbitrary units – AUI) has been normalized to the corresponding (per condition) cell density (Janus Green staining). Each bar represents the mean±SEM from five independent experiments. Data were processed with two-way ANOVA, followed by post hoc Bonferroni’s multiple comparison t-test: ***p≤0.001 compared to the control (CNTRL; unstressed/untreated conditions); #p≤0.05 & ###p≤0.001 compared to the TG-stressed neurons; ∂∂p≤0.01 compared to the Liraglutide-treated, TG-stressed neurons.
Liraglutide co-treatment significantly ameliorates the suppressed basal respiration (two-way ANOVA interaction: F (1,11) = 15.38, p = 0.0024), maximal respiration (two-way ANOVA interaction: F (1,11) = 5.497, p = 0.0389) and spare respiratory capacity (two-way ANOVA interaction: F (1,11) = 3.55, p = 0.05) in chronically thapsigargin-stress neurons (Fig. 2a, b). It attenuates the reduced basal glycolytic rate (two-way ANOVA main effect: F (1,11) = 9.33, p = 0.011), basal PER (two-way ANOVA main effect: F (1,11) = 8.36, p = 0.015), and compensatory glycolytic rate (two-way ANOVA main effect: F (1,11) = 10.24, p = 0.0085) (Fig. 3). The latter parallels the fold decrease in the aggregated-over-monomer JC1 ratio, as respectively shown in Fig. 4a, that suggests an amelioration of the perturbed mitochondrial polarization.
The dual incretin similarly re-initiates the arrested oxidative phosphorylation (two-way ANOVA interaction of dual agonist×thapsigargin: basal OCR, F (1,12) = 10.18, p = 0.0078; maximal OCR, F (1,12) = 8.045, p = 0.0150; spare OCR, F (1,12) = 5.552, p = 0.0363) (Fig. 2) and glycolysis (two-way ANOVA main effect: basal glycolysis, F (1,11) = 10.83, p = 0.0072; basal PER, F (1,11) = 19.47, p = 0.001; compensatory glycolysis, F (1,11) = 12.52, p = 0.0046) (Fig. 3) in the thapsigargin-treated neurons. At the same time, it reduces by more than half the ratio of the aggregated to the monomeric forms of JC1, corresponding to an approximate normalization of the values. Notably, the co-stimulation of GLP-1 and GIP receptors maximizes the beneficial effect of the incretin treatment for the regulation of the ΔΨm under conditions of persistent ER stress (two-way ANOVA interaction: F (1,86) = 9.931, p = 0.0022), as illustrated in Fig. 4a, which may further underlie the trend of the superior effect in oxidative metabolism and anaerobic cycling of glucose, as noted in Figs. 2b and 3b.
Mechanistically, the homeostasis of the mitochondrial membrane permeabilization is crucial for cell fate, functional fidelity of the electron transport chain for mitochondrial respiration, and the import of metabolites in the mitochondrial lumen. All these functions integrate into the availability and activation patterns of selected Bcl-2-family members, such as Bcl-2, BAD, and BID [36, 37]. With these regards, we have next quantified functional phosphorylation of Bcl-2 at the serine 70 [34] residue and the expression of the full-length BID by western blotting (Fig. 4b, c). Chronic ER stress with thapsigargin halves the expression levels of the phosphorylated and total Bcl-2 that signal for a notable inhibition of this protein. It further downregulates the expression of full-length BID by 70%, indicative of its truncation into the active pro-apoptotic form [35]. ELISA-based protein microarray additionally revealed that persistent SERCA favors the de-phosphorylation of BAD (Fig. 8d) that, in turn, can bind and inhibit the pro-survival molecules BclxL, Bcl-2, and Bcl-W [36, 37].
Liraglutide co-treatment ameliorates the aberrant inhibition of Bcl-2 under conditions of irreversible ER stress (two-way ANOVA interaction: F (1,28) = 4.573, p = 0.0413). The GLP-1/GIP dual agonist maximizes the beneficial effect of incretin co-treatment on the function and availability of this Bcl-2-family member for survival signaling and oxidative phosphorylation (two-way ANOVA interaction of dual agonist×thapsigargin: F (1,28) = 17.93, p = 0.0002; two-way ANOVA interaction of GIP-receptor co-stimulation×thapsigargin vs. GLP-1 mono-stimulation: F (1,28) = 8.498, p = 0.0069), as shown in Fig. 4b. Furthermore, incretin co-treatments normalize the suppressed expression of the full-length BID (two-way ANOVA interaction of Liraglutide×thapsigargin: F (1,28) = 23.50, p < 0.0001; two-way ANOVA interaction of dual agonist×thapsigargin: F (1,28) = 19.47, p = 0.0001), as illustrated in Fig. 4c. Incretin co-treatments additionally restore the aberrant phosphorylation of BAD at the serine 112 residue (two-way ANOVA interaction of Liraglutide×thapsigargin: F (1,28) = 15.98, p = 0.0004; two-way ANOVA interaction of dual agonist×thapsigargin: F (1,28) = 5.985, p = 0.0210) back to basal levels (Fig. 8d).
Next, we explored the hypothesis of whether the incretin-driven restoration of the mitochondrial respiration could have been partially attributed to the enhanced mitochondrial biogenesis under ER stress conditions. For this purpose, we quantified the expression ratio of the main subunit of complex IV, COX1 over the subunit A of respiratory complex II, SDH-A with a kinetic ELISA-based assay, as illustrated in Fig. 5a. We initially observed that the chronic thapsigargin treatment detrimentally affects chromophore development over time (three-way ANOVA interaction: F (13,975) = 23.74, p < 0.0001) and renders a decrease at the endpoint values of the ratio by 30% (Fig. 5b). This finding signifies suppressed mitochondrial biogenesis under conditions of persistent ER stress.
Fig. 5
Liraglutide and the GLP-1/GIP dual agonist mitigate the impaired mitochondrial biogenesis following irreversible ER stress. On d 6 of the differentiation period, dopaminergic-like neurons from the human LUHMES cell line were treated with 0 and 100 nM of thapsigargin (TG) in the presence or absence of each incretin tested for 16 h. Neurons were fixed with 4% paraformaldehyde and monitored for the protein expression ratio of the cytochrome c oxidase subunit I (COX1; mitochondrial DNA-encoded protein) to the succinate dehydrogenase (SDH-A; nuclear DNA-encoded protein) over a kinetic cycle of 15 min for signal development with a colorimetric ELISA assay (a). Endpoint measurements of the ratio are expressed as percentages of the control (CNTRL; untreated/unstressed conditions) and illustrated as a bar graph (b). Each point and bar represent the mean±SEM from five independent experiments. Data processed with two-way ANOVA, followed by post hoc Bonferroni’s multiple comparison t-test: **p≤0.01 compared to the control (CNTRL; unstressed/untreated conditions); #p≤0.05 & ##p≤0.01 compared to the TG-stressed neurons.
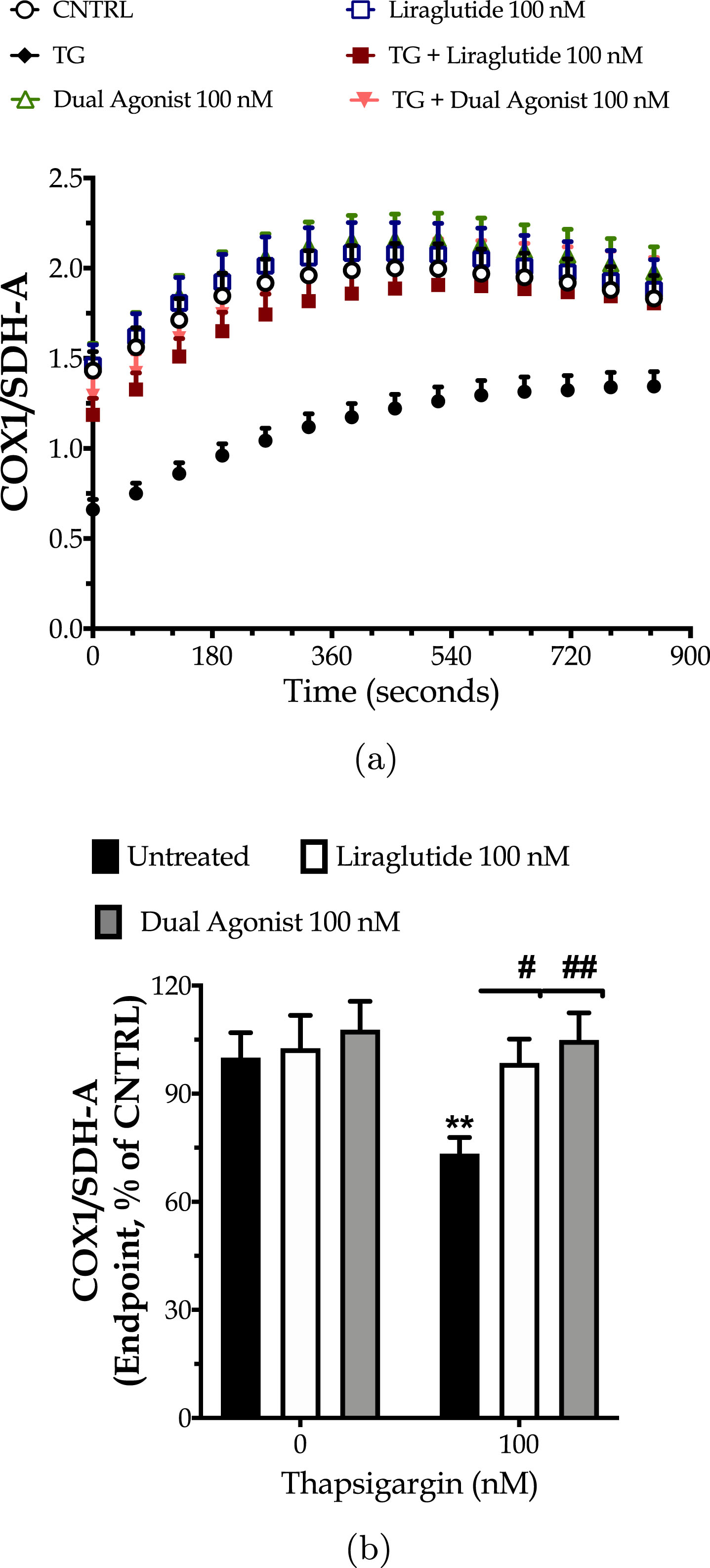
Incretin co-treatments significantly improve the signal development over time (three-way ANOVA interaction of time×thapsigargin×Liraglutide: F (13,975) = 2.065, p = 0.0140; time×thapsigargin×dual incretin: F (13,975) = 4.151, p < 0.0001) that corresponds to a normalization of the expression ratio of the mitochondrial over the nuclear DNA-encoded complex subunits, as shown in Fig. 5. Taken together, our findings pinpoint how beneficial is the engagement of GIP and/or GLP-1 receptors in maintaining metabolically-active neuronal cells in times of need.
Incretin co-treatments mitigate the autophagy arrest.
We have previously reported that the chronically persistent SERCA channel inhibition diminished the availability of components crucial for autophagosome formation and maturation and thus precluded macroautophagy (hereinafter called as autophagy) in the neuroblastoma SH-SY5Y [27] and hepatoma HepG2 [30] cell lines. Similarly, herein, we have observed that chronic thapsigargin treatment halves the neuronal expression of the autophagy-related (Atg) proteins beclin-1, Atg3, Atg7, and LC3B (Fig. 6) that sequentially regulate the initiation, elongation and maturation of the autophagosome [10].
Fig. 6
Liraglutide (LIRA) and the GLP-1/GIP dual agonist (DA) normalize the suppressed expression of core autophagy-related proteins (Atg) proteins for autophagosome formation and maturation upon chronic ER stress. On d 6 of the differentiation period, dopaminergic-like neurons from the human LUHMES cell line were treated with 0 and 100 nM of thapsigargin (TG) in the presence or absence of each incretin tested for 16 h. Neurone were harvested, and the expression of beclin-1 (a), Atg3 (b), Atg7 (c) and of LC3 (d) were determined by western blotting. β-Actin was used as the loading control in our quantification. Loading controls for a and c are the same. Each bar represents the mean±SEM from five independent experiments. Data expressed as fold change to the control (CNTRL; unstressed/untreated conditions) and processed with two-way ANOVA, followed by post hoc Bonferroni’s multiple comparison t-test: ***p≤0.001 compared to CNTRL; ###p≤0.001 compared to the TG-stressed neurons.
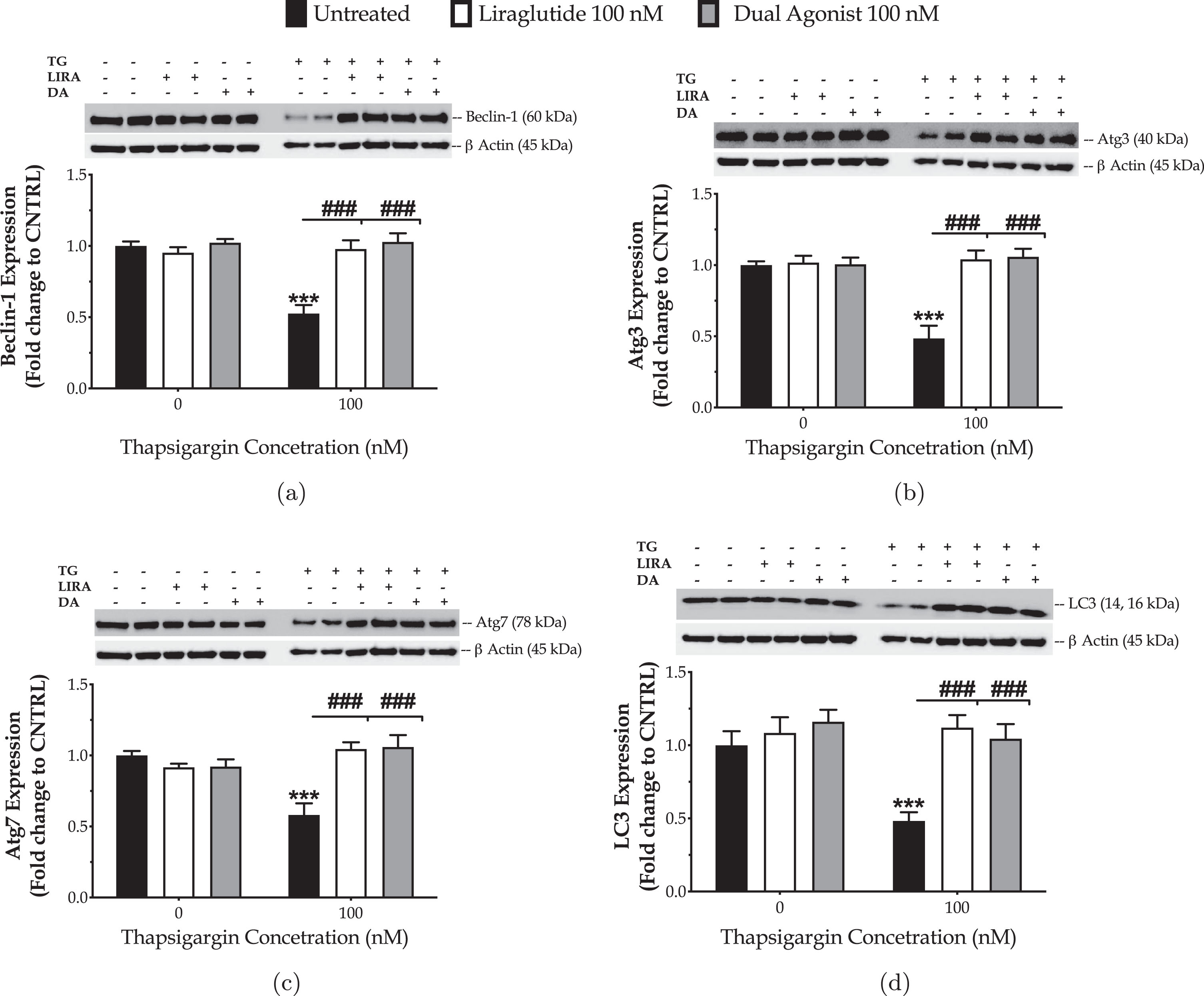
Liraglutide co-treatment restores the suppressed expression of beclin-1 (two-way ANOVA interaction: F (1,28) = 25.76, p≤0.001), Atg3 (two-way ANOVA interaction: F (1,28) = 19.69, p = 0.0001), Atg7 (two-way ANOVA interaction: F (1,28) = 28.21, p≤0.001), and LC3 (two-way ANOVA interaction: F (1,28) = 9.815, p = 0.004), as illustrated inFig. 6.
Similarly, the dual incretin normalizes the aberrant protein levels of beclin-1 (two-way ANOVA interaction: F (1,28) = 25.68, p≤0.001), Atg3 (two-way ANOVA interaction: F (1,28) = 23.14, p≤0.001), Atg7 (two-way ANOVA interaction: F (1,28) = 17.86, p = 0.0002), and LC3 (two-way ANOVA interaction: F (1,28) = 5.483, p = 0.0265) in the neurons subjected to chronic ER stress (Fig. 6). No superior efficacy of the dual incretin in regulating the expression of the components of the autophagic machinery was noted, when compared to Liraglutide (three-way ANOVA interaction of Atg proteins×thapsigargin×GIP-receptor co-stimulation: F (3,112) = 0.3943, p = 0.7574).
Incretin co-treatments allay the synaptic and neuronal cell death
Followingly, we asked how rectifying the aberrant ER-mitochondrial-autophagic axis could have affected synaptic and neuronal-cell fate. For this purpose, we quantified the expression levels of the hallmark scaffolding protein for dendritic spine morphogenesis and synaptic plasticity, PSD95 and of the major pre-synaptic vesicle protein, synaptophysin by immunoblotting. We further quantified the metabolization of XTT into the orange, water-soluble formazan product and the content of the LDH in the culture supernatant to assess cell viability and cytotoxicity, respectively. As evident in our findings in Fig. 7a and 7b, Liraglutide restores the suppressed expression of the PSD95 (two-way ANOVA interaction: F (1,28) = 23.493, p < 0.0001) and synaptophysin markers (two-way ANOVA interaction: F (1,28) = 6.83, p = 0.0142). The biochemical observations are accompanied by a significant amelioration of the thapsigargin-driven neuronal-cell death (two-way ANOVA interaction for XTT: F (1,92) = 5.343, p = 0.023; two-way ANOVA interaction for LDH: F (1,108) = 10.91, p = 0.0013) (Fig. 7c, d).
Fig. 7
Incretin co-treatments allay the synaptic and neuronal-cell death upon irreversible ER stress. On d 6 of the differentiation period, dopaminergic-like neurons from the human LUHMES cell line were treated with 0 and 100 nM of thapsigargin (TG) in the presence or absence of each incretin tested for 16 h. Neurons were then harvested, and the expression of postsynaptic density protein 95 [PSD95; (a)] and synaptophysin [Sys; (b)] were determined by western blotting. β-Actin was used as the loading control in our quantification. Alternatively, neurons were assayed for the XTT metabolization (c) to assess cell viability. Spent supernatant was collected and processed for the LDH activity (d) to determine necrosis. Each bar or line represents mean±SEM from five independent experiments. Data are expressed as fold change to the control (CNTRL; unstressed/untreated conditions) and processed with two-way ANOVA, followed by post hoc Bonferroni’s multiple comparison t-test: **p≤0.01 & ***p≤0.001 compared to CNTRL; #p≤0.05, ##p≤0.01 & ###p≤0.001compared to the TG-stressed neurons.
![Incretin co-treatments allay the synaptic and neuronal-cell death upon irreversible ER stress. On d 6 of the differentiation period, dopaminergic-like neurons from the human LUHMES cell line were treated with 0 and 100 nM of thapsigargin (TG) in the presence or absence of each incretin tested for 16 h. Neurons were then harvested, and the expression of postsynaptic density protein 95 [PSD95; (a)] and synaptophysin [Sys; (b)] were determined by western blotting. β-Actin was used as the loading control in our quantification. Alternatively, neurons were assayed for the XTT metabolization (c) to assess cell viability. Spent supernatant was collected and processed for the LDH activity (d) to determine necrosis. Each bar or line represents mean±SEM from five independent experiments. Data are expressed as fold change to the control (CNTRL; unstressed/untreated conditions) and processed with two-way ANOVA, followed by post hoc Bonferroni’s multiple comparison t-test: **p≤0.01 & ***p≤0.001 compared to CNTRL; #p≤0.05, ##p≤0.01 & ###p≤0.001compared to the TG-stressed neurons.](https://content.iospress.com:443/media/jpd/2023/13-7/jpd-13-7-jpd230030/jpd-13-jpd230030-g007.jpg)
Similarly, the dual incretin reverts the deficient availability of PSD95 (two-way ANOVA interaction: F (1,28) = 33.40, p < 0.0001), and synaptophysin (two-way ANOVA interaction: F (1,28) = 4.661, p = 0.0396) to the control levels upon chronic thapsigargin co-treatment. It further hinders the exacerbated LDH release into the supernatant (two-way ANOVA interaction: F (1,101) = 9.563, p = 0.0026) that is indicative of constraining aberrant necrosis driven by persistent ER stress (Fig. 7d). The latter correlates with an amelioration of the decreased XTT metabolization (two-way ANOVA interaction: F (1,92) = 25.09, p < 0.0001), which further confirms that incretin-cotreatment switches neuronal towards survival under conditions of persistent ER stress (Fig. 7c). No statistically superior efficacy of the dual incretin over Liraglutide was observed, though a trend of better regulation of synaptic and cell-viability markers is prominent in Fig. 7.
Incretin co-treatments reengage STAT3, Akt, and Nrf2 signaling to preclude neuronal degeneration
Previously, we have reported that the mono-activation of GLP-1 receptor re-instates homeostasis of the signal transducer and activator of transcription 3 (Stat3) and Akt signaling pathways to pave the way for normal cell proliferation and viability following chronic ER stress in the neuroblastoma SH-SY5Y cell line [27]. Herein, we employed the same ELISA-based protein microarray (Supplementary Figure 1) to confirm/examine whether the incretin co-treatments would regulate the same intracellular pathways in human dopaminergic-like neurons under conditions of chronic perturbation of the ER-mitochondrial-autophagic axis (Fig. 8 and Supplementary Table 2).
Fig. 8
Incretin co-treatments restore the derailed Akt and Stat3 intercellular signaling to halt the ectopic poly (ADP-ribose) polymerase (PARP) and caspase 3 (CASP3) cleavage, balance the expression and activation of the pro-survival Bcl-2 over the pro-apoptotic BID, and thus to pave the way for survival of the apoptotic neuron subjected to chronic ER stress. On d 6 of the differentiation period, dopaminergic-like neurons from the human LUHMES cell line were treated with 0 and 100 nM of thapsigargin (TG) in the presence or absence of each incretin tested for 16 h. Neurons were then harvested, and 0.3 mg mL1 protein of whole-cell lysate was processed with the PathScan® sandwich immunoassay. All protein samples per experiment were processed in duplicate. Each bar represents the mean±SEM from five independent experiments. Data are expressed as fold change to the control (CNTRL; unstressed/untreated conditions) and analyzed by two-way ANOVA, followed by post hoc Bonferroni’s multiple comparison t-test: *p≤0.05, **p≤0.01 & ***p≤0.001 compared to CNTRL; #p≤0.05, ##p≤0.01 & ###p≤0.001, & nsp»0.05 compared to the TG-treated neurons.
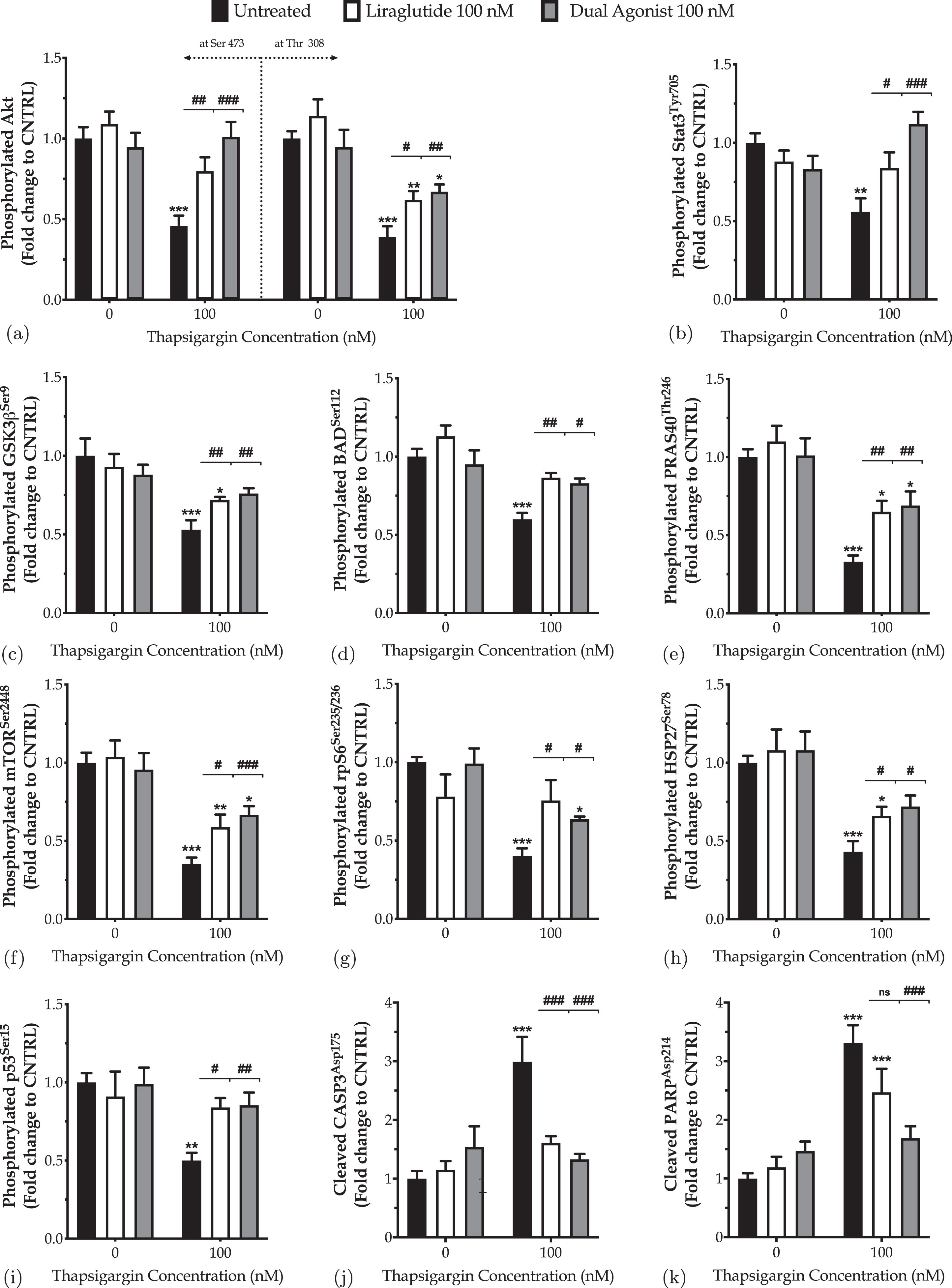
Both Liraglutide and dual incretin mitigate the suppressed expression of the phosphorylated Akt at the serine 473 and threonine 308 residues (Fig. 8a) and of the activating phosphorylation of STAT3 at the tyrosine residue 705 (Fig. 8b) in differentiated LUHMES cells chronically challenged with 100 nM of thapsigargin. These findings are accompanied by relieving the thapsigargin-driven inhibition of the activating phosphorylation of the downstream proline-rich Akt substrate of 40 kDa (PRAS40), mammalian target of rapamycin (mTOR), and ribosomal protein S6 (rpS6) kinases at Thr246, Ser2448, and Ser235/236, respectively (Fig. 8e–g).
Besides, chronic ER stress engages the glycogen synthase kinase 3β (GSK3β) (Fig. 8c) by halving the inhibitory phosphorylation of the kinase at the serine 9 residue (p≤0.001). In contrast, it impedes the phosphorylation of the heat shock protein 27 (HSP27) (Fig. 8h) and stress-responsive p53 (Fig. 8i) at Ser78 (p≤0.001) and Ser15 (p≤0.01), respectively. Liraglutide restores the expression of the phosphorylated p53 back to control levels. It significantly ameliorates the suppressed phosphorylation of HSP27 and GSK3β, though their levels have remained significantly decreased when compared to control conditions (p≤0.05). The dual incretin co-treatment normalizes the derailed GSK3β (Fig. 8c), p53 (Fig. 8i) and HSP27 (Fig. 8h) activity under chronic thapsigargin treatment, signifying for a possible superior efficacy of the dual incretin over Liraglutide in rectifying intracellular signaling upon chronic ER stress in dopaminergic-like neurons.
Furthermore, the incretin-induced alleviation of the perturbed intracellular signaling was accompanied by a significant inhibition of proteolytic cleavage of caspase 3 (CASP3) (Fig. 8j) and its downstream nuclear enzyme poly (ADP-ribose) polymerase (PARP) (Fig. 8k), which both function as critical executioners of cell death [29]. Specifically, Liraglutide co-treatment restores the expression of the cleaved active fragment of CASP3 back to control levels (p≤0.001), though it leaves unaffected the levels of cleaved PARP under chronic ER stress conditions. Intriguingly, the dual-incretin co-treatment normalizes the ectopic proteolytic cleavage of CASP3 (Fig. 8j) and PARP (Fig. 8k) following chronic thapsigargin treatment; this finding signals for the necessity of the GIP receptor co-stimulation in mitigating death signals under the apoptotic phase of UPR in dopaminergic-like neurons.
Finally, we performed immunocytochemical analysis of the nuclear levels of the nuclear factor erythroid-derived 2-like 2 (Nrf2) to evaluate the antioxidant defensive signaling for neuroprotection upon aberrant proteostasis following chronic thapsigargin treatment (Fig. 9). Normally, Nrf2 is expressed throughout the neuron soma with a predominant expression in the nucleus [38], as prominent in our immunocytochemical analysis too (Fig. 9a). Contrarily, degenerating neurons feature a decreased nuclear Nrf2 content that signifies the impaired activity of the transcription factor [38]. Accordingly, herein, thapsigargin-treated neurons display a faint Nrf2-immunopositive staining, particularly in the nuclear area (Fig. 9a). One-way ANOVA analysis demonstrated significant differences in the nuclear Nrf2 content among the groups (F (5,192) = 4.635, p≤0.001). Two-way ANOVA analysis revealed that Liraglutide (F (1,122) = 11.57, p≤0.001) and dual-incretin (F (1,128) = 17.30, p≤0.001) co-treatments significantly modulate the Nrf2 nuclear content and notably normalize it, as illustrated in Fig. 9b.
Fig. 9
Liraglutide (LIRA) and the GLP-1/GIP dual agonist (DA) reinstates homeostasis of the attenuated signaling of the nuclear factor erythroid 2-related factor 2 (Nrf2) upon irremediable ER stress. On d 6 of the differentiation period, dopaminergic-like neurons from the human LUHMES cell line were treated with 0 and 100 nM of thapsigargin (TG) in the presence or absence of each incretin tested for 16 h. All cell treatments were performed in duplicate per experiment. Neurons were (para-formaldehyde)-fixed, immunolabelled for Nrf2 and processed for confocal imaging at 40X magnification, as shown in the representative images (a). Six pictures were captured per treatment sample per experiment for quantification. Image J was used to quantify corrected total cell fluorescence (CTCF) of the nuclear Nrf2 staining (b). Each bar represents the mean±SEM from five independent experiments. Data were processed with two-way ANOVA, followed by post hoc Bonferroni’s multiple comparison t-test: ***p≤0.001 compared to CNTRL; ##p≤0.01 & ###p≤0.001 compared to the TG-stressed neurons. Scale bars: 50μm.
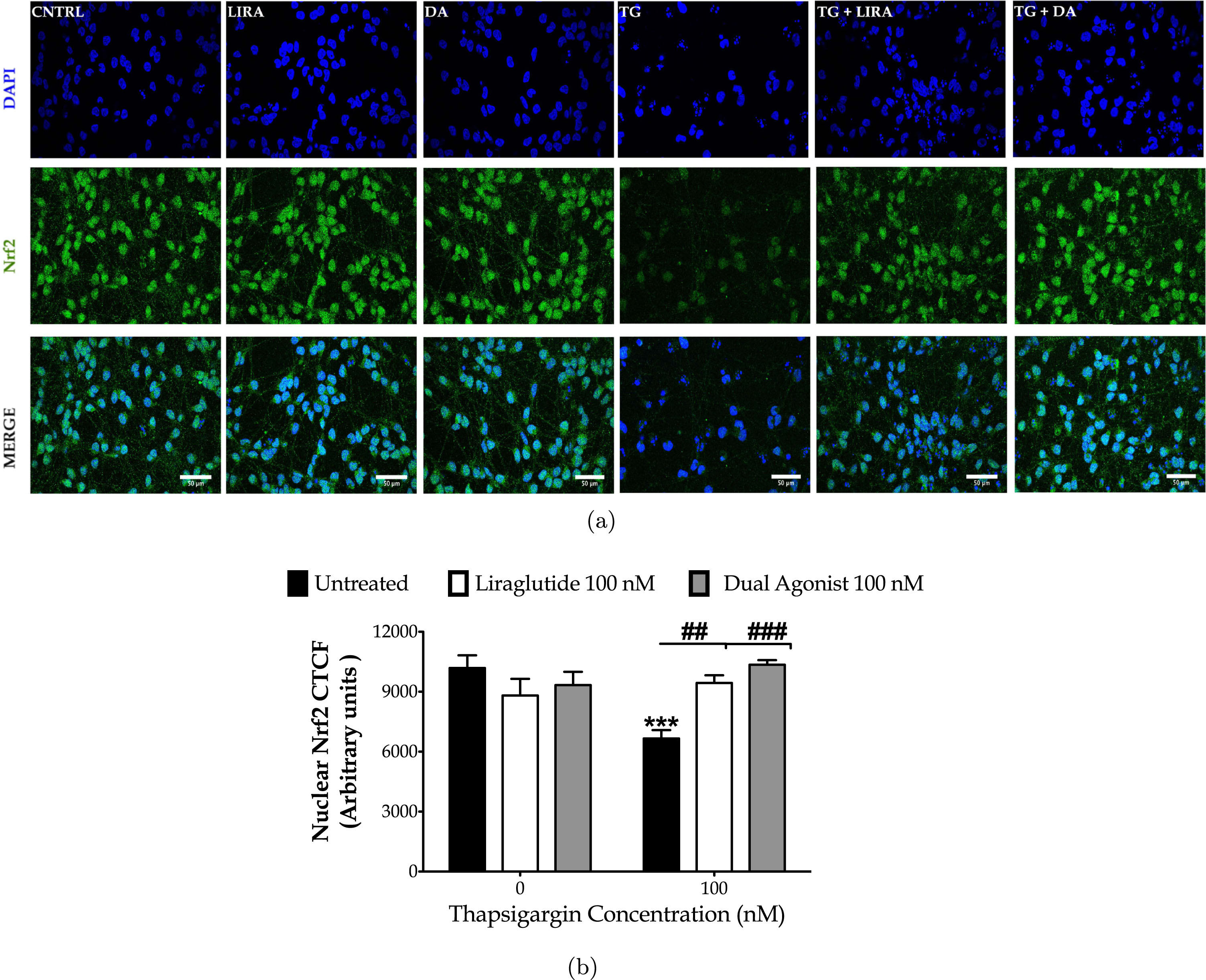
Fig. 10
Summarizing illustration for incretin-driven effects and signaling observed upon chronic ER stress in dopaminergic-like neurons from the human LUHMES cell line. Liraglutide and GLP-1/GIP dual agonist modulate the effector Akt and Stat3 signaling to regulate the activity and expression patterns of downstream pro-apoptotic proteins (in black), pro-survival factors (in light red) and critical signaling pathways (in green). In turn, they restore essential intracellular processes to ultimately elicit neuroprotection and synaptic repair. For abbreviations, please refer to the main text.
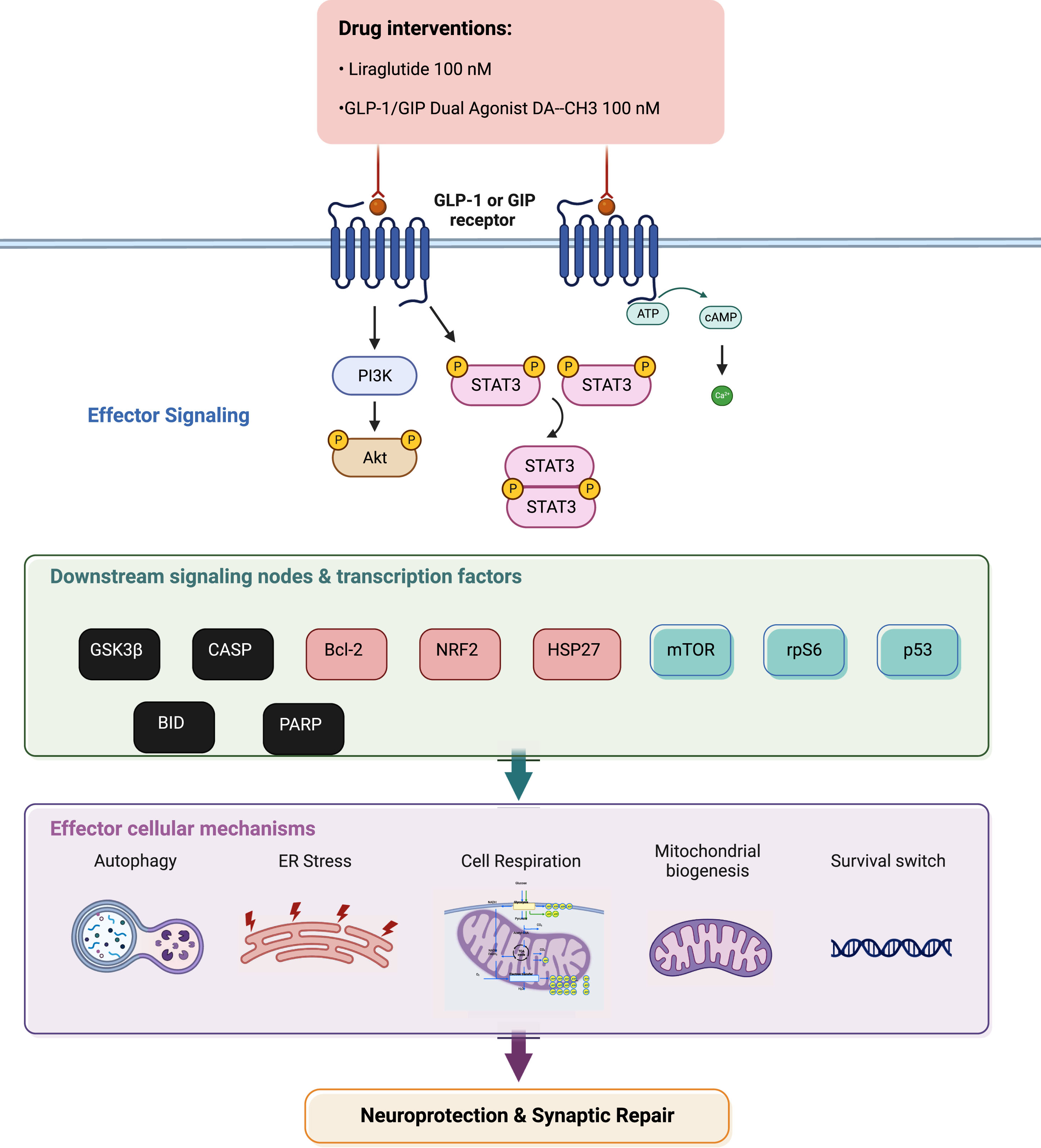
DISCUSSION
GLP-1 receptor agonists have shown neuroprotective effects in a number of clinical trials. The drug exenatide (exendin-4) showed clear protective effects in PD patients [21, 40], and a phase 3 testing this drug is currently ongoing [22]. A phase 2 clinical trial testing Liraglutide showed similar neuroprotective effects in PD patients [23], demonstrating that this drug target is viable and can make meaningful changes in disease progression. Liraglutide furthermore showed good effects on cognition and brain shrinkage in a phase 2 clinical trial in AD patients [24]. However, our knowledge of the underlying mechanisms of GLP-1 and GIP signaling in neurons is limited [39], and the current study adds and confirms important molecular processes that play a key part. Chronic ER stress has emerged as a hub for the derailed proteostasis underlying the onset and progression of neuronal degeneration, with the UPR and autophagy deregulation paralleling the temporal and spatial pattern of the pathologic protein deposition in human brain [40–43]. Chronic ER stress disproportionately impacts the signaling dynamics of the PERK, ATF6, and Ire1α branches to determine cell fate [44, 45]. It suppresses ATF6 and Ire1α signaling [45, 46] to possibly attenuate the downstream survival signals and the neurotrophic effects of the spliced X-box binding protein 1 (XBP-1) [41]. Spliced XBP1 regulates the transcription of the brain-derived neurotrophic factor (BDNF) and other genes related to BDNF signaling to fine-tune synaptic plasticity in the murine hippocampus [47]. Accordingly, we show that chronic perturbation of ER calcium diminishes the phosphorylation of Ire1α at Ser724 and the expression of ATF6 that correlate to a decrease in the plasticity-related proteins, PSD95 and synaptophysin.
Additionally, Ire1α activation is a prerequisite for initiating pro-survival autophagy in response to ER stress [48]. It mediates the phosphorylation of Bcl-2 to favor the dissociation of beclin-1 from its inhibitory complex with Bcl-2 at the ER membrane [49]. It also stimulates the transcription factor XBP1, which, in turn, bounds to the promoter of beclin-1 and induces its transcription [50]. Herein, we show that the dopaminergic-like neurons feature the suppressed Ire1α and Bcl-2 activity following irremediable ER stress that may account for the observed beclin-1 deficiency. For instance, a substantial loss of beclin-1 has been detected in the mid-frontal cortex of AD patients [51] and cingulate cortex of PD patients [52] Genetic depletion of beclin-1, furthermore, exacerbates amyloid-β (Aβ) pathology and dampens the expression of synaptophysin, the dendrite-specific microtubule- associated protein 2 (MAP2) and of the calcium-binding protein, calbindin to potentiate synapse degeneration in vivo [51]. Whilst, gene transfer to enhance beclin-1 expression has been shown to reduce α-synuclein accumulation in α-synuclein-overexpressing mice [53] and PC12 [54] cells.
Contrarily to Ire1α, chronic ER stress augments the activity of PERK to amplify and sustain the expression of the short-lived, pro-apoptotic transcription factor Chop [45]. Chop inhibits the expression of the pro-survival Bcl-2 factor whilst stimulating the transcription of the Ero1-Lα to provoke the mitochondrial apoptotic machinery and neuronal damage [42, 44], as prominent in our findings too. Ero1-Lα harnesses the oxidizing power of the molecular oxygen to oxidize the active cysteinyl-thiol groups in PDI, enabling the latter to introduce disulfide bonds to folding substrates and to maintain the thiol-disulfide exchange and PDI capacity for the bond isomerization within nascent polypeptides [55]. The redox fidelity of the oxidoreductase and the detoxification of the hydrogen peroxide produced upon the formation of a disulfide bond relies on the glutathione metabolism [55] that is contingent on the Nrf2 activity [56]. Herein, we show the Ero1-Lα excess correlates to PDI deficiency and a decreased Nrf2 nuclear (active) content, signaling for defective oxidative folding and high ER content of hydrogen peroxide. The latter along with the observed downregulation of calnexin can exacerbate the abnormal protein load within the organelle lumen and thus the ER stress magnitude to hasten cell demise [57, 58]. In addition, high levels of hydrogen peroxide drive the activation of the inositol-1,4,5-trisphosphate receptor (IP3R) and increase the levels of calcium on the cytosolic face of the ER. The latter along with the deficiency of calnexin that normally regulates the retrieval of the release calcium to the ER perturbs the calcium availability into the mitochondrion [59]. Aberrant calcium dynamics limit mitochondrial respiration, whilst inducing the core apoptosis machinery [60] and the proteolytic (activating) cleavage of CASP12 [61]. Active CASP12 potentiates a downstream caspase cascade (e.g., CASP3) that mediates synaptophysin depletion and executes PARP degradation to assure the fulfilment and irreversibility of the apoptotic process for synapse and neuronal toxicity [62–66], as reflected in our results too. Besides the modulation of redox signaling and core apoptosis machinery, Chop can obscure autophagy through the transcriptional regulation of Atg components for phagophore elongation and maturation into the autophagosome, including the Atg7 gene [67, 68]. Atg7 deficiency potentiates a spontaneous accumulation of protein aggregates, neuronal degeneration, and lossin vivo [69].
Liraglutide and dual incretin co-treatments both alleviate the ectopic BiP and Chop expressions, rectify the aberrant Ire1α, Nrf2 and CASP12 activity, and restore the protein levels of ATF6, PDI, calnexin, Ero1-Lα, beclin-1, Atg3, Atg7, and LC3. These biochemical traits correspond to the amelioration of the suppressed oxidative phosphorylation and glycolysis and mitochondrial hyperpolarization. They additionally correlate with the normalization of PSD95 and synaptophysin expression and restraint of the aberrant neuronal toxicity in the XTT-assessed metabolic activity assay, LDH levels and the proteolysis of the executioner CASP3 and PARP. Our results collectively suggest that the incretin mimetics shift the ‘terminal’ signals of UPR into adaptive that favors the homeostasis of autophagy, mitochondrial and quality control machineries. This in turn abets the proteostasis, cell bioenergetics, and neuronal repair following persistent ER stress, with the GIP receptor co-stimulation maximizing the neuroprotective effect of the GLP-1 receptor.
Mechanistically, the incretin mimetics restore the suppressed STAT3 and Akt signaling to elicit neuroprotection upon persistent ER stress. STAT3 is a latent transcription factor that lies downstream the stimulation of cytokine and growth factor receptors, including the GLP-1 receptor [70]. Suppressed STAT3 activity occurs in the hippocampus of affected patients and animals of AD [71]. Chiba et al. have demonstrated that Aβ inactivates the hippocampal JAK2 (Janus kinase 2)/STAT3 axis to compromise basal forebrain cholinergic function and induce spatial working memory deficits in vivo, linking the STAT3 pathway to neurodegenerative processes [71]. Subsequent work has further revealed that the ER stress perturbs the astrocytic STAT3 signaling to modulate acute-phase neuroinflammatory responses in a PERK-dependent fashion [66]. Although the mechanistic interplay between the UPR and STAT3 signaling in the context of neuronal functioning and fate remains elusive, STAT3 regulates the transcription of the pro-survival Bcl-2 protein and axonal outgrowth/branching programs to promote neuronal survival and axonal regeneration post oxygen-glucose deprivation and excitotoxicity [72, 73]. Activated STAT3 additionally induces the expression of synaptophysin that enhances synaptogenesis and synaptic plasticity in the hippocampus and cortex [72, 74]. It further regulates the expression of nuclear-encoded genes for oxidative phosphorylation [75], e.g., subunits of Complex I and V, and the expression of the citrate synthase – the rate-limiting enzyme for the induction of the Krebs’s cycle [76] to determine cell metabolism functioning and efficiency [75, 76]. It can modulate HIF-1α transcriptional response to promote glycolysis and regulate metabolic switch in times of need [77, 78]. Taken together, the restoration of STAT3 activation serves as important signaling node through which GLP-1 and GIP receptors can confer trophic signals for the synapse homeostasis, mitochondrial biogenesis, resolution of respiratory arrest, and neuronal repair/adaptation following chronic ERstress.
Akt is a serine/threonine kinase with a wide spectrum of targets in the cytoplasm, nucleus, mitochondria, and the ER membrane to regulate adaptive responses and cell fate under stress conditions [79, 80]. Herein, we show Liraglutide and the dual incretin alleviate the ER stress-induced inhibition of Akt phosphorylation at Thr308 and Ser473 sites, signaling for rescuing its maximal activity [79]. The latter corresponds to an amelioration of the suppressed phosphorylation of PRAS40 at Thr246 and thus relieves the PRAS40 inhibition of the mTOR signaling [81]. The importance of this observation lies on the fact that the Akt/mTOR network function as a sensor for integrating intracellular and extracellular cues to modulate an array of basic cellular processes, including protein synthesis, ribosomal biogenesis, autophagy, lipid and nucleotide synthesis, and bioenergetics, to orchestrate adaptive responses to stress and nutrient disturbance [80, 82, 83]. ER stress derails mTOR activity and renders neurons vulnerable to Chop/Ero1-Lα-mediated oxidative damage; as such it facilitates the intrinsic apoptotic machinery [84], as reflected in our findings too. Suppressed mTOR has been previously reported to underlie the downregulated expression of the activity-regulated cytoskeleton-associated protein (Arc) for synaptic plasticity and the impaired long-term potentiation in degenerating murine hippocampus [85]. Suppressed mTOR further abolishes the inhibitory phosphorylation of Atg13 and unc-51-like kinase 1 (ULK1) that allow the formation of the ULK complex and the induction of autophagy [83, 85]. However, excessive autophagic flux degrades endogenous inhibitors of apoptosis and Atg components and detrimentally affects cell fate[84, 85].
Furthermore, Akt phosphorylates and inhibits GSK3β that critically drives neuronal apoptosis following ER stress [27, 86–89]. In the degenerating brain, GSK3β immunoreactive granules occur in neurons with activated PERK signaling [90, 91]. Activated PERK engages GSK3β activity through selective removal of the inactive (phosphorylated at Ser9) kinase form via the autosomal-lysosomal pathway [90]. GSK3β, in turn, inhibits Nrf2 by nuclear exclusion and proteasomal degradation [80, 83]. The latter could preclude the availability of substrates and oxidative-phosphorylation subunits for respiration and ATP formation, critical enzymes for fatty acid synthesis and glutathione cycle regulation, and mitochondrial membrane potential [92]. Active (nuclear) Nrf2 also induces the expression of regulatory proteins for the autophagy initiation, cargo recognition, autophagosome formation, elongation and autolysosome clearance [56, 93] whilst precluding the recruitment of ATF4 to the Chop promoter and thereby the Chop transcription[94].
In addition, Akt phosphorylates and inhibits the death-agonist BAD, which becomes rapidly de-phosphorylated upon apoptotic stimuli [80]. The restoration of BAD phosphorylation, through Akt, raises the mitochondrial threshold for apoptosis and renders cells less vulnerable to death signals [95], as prominent in our results too. Akt can further phosphorylate the stress-responsive HSP27 [96] that sequesters cytochrome c and/or pro-caspase 3 to preclude the apoptosome formation and downstream death signal transduction [97–99]. Furthermore, activated HSP27 phosphorylates p53 at Ser15 and induces its downstream target gene expression to prevent genototoxicity [97, 100]. Lastly, activated Akt may also phosphorylate and inhibit PERK kinase [101], offering an additional mechanistic link for the neuroprotective and restorative effects of incretin mimetics upon persistent ER stress. Exacerbated PERK activity triggers general translation repression and downregulation of the plasticity-related cyclic AMP response-element-binding protein (CREB), through its downstream targets eIF2α and ATF4 respectively, to provoke PSD95 deficiency, synapse damage and cognitive decline in vivo [42]. Although we have not assessed the expression of phosphorylated PERK levels, transcriptional and translational regulation of Chop expression primarily lies downstream of the PERK arm [45]. Taken together, our findings suggest that the stimulation of GIP and/or GLP-1 receptors rectify the Akt activation to counteract the ER stress-driven suppression of mTOR and HSP27 signaling and to hamper the exacerbated GSK3β activity. These effects, in turn, normalize Nrf2 nuclear levels and reinforce trophic signals and mitochondrial function upon chronic ER stress for neuronal survival and availability of the major synaptic proteins, PSD95 and synaptophysin. Synapse degeneration and loss plays a major role in disease progression of AD and PD, and incretins have been shown to re-activate synaptic activity and plasticity in the brain [46].
Though not addressed in the present study, incretin mimetics may have resolved the ER stress and elicited neuroprotection through direct modulation of intracellular calcium fluxes. Previous studies have demonstrated that the stimulation of GLP-1 receptor potentiates the formation of cyclic AMP to regulate calcium responses and thus rescue hippocampal neurons and SH-SY5Y cells from apoptosis driven by excitotoxicity and oxidative stress [102–104]. As such, future studies should examine the effect of incretin mimetics in SERCA channel expression and activation patterns following chronic ER stress with thapsigargin. Furthermore, herein, we show that the ER stress and UPR deregulation coincides with deficiency of critical Atg components. Among the proteins monitored, we quantified the expression of the LC3 protein that functions in the autophagosome biogenesis [40]. The lack of detection of two district bands in our immunoblotting studies has precluded the quantification of the ratio of the lipidated and autophagosome-recruited type II versus the cytosolic type I of LC3 for the autophagy flux assessment. Electron microscopy is the most sensitive technique for the identification, morphological characterization, and visualization of autophagic structures in their complex physiological environment [105]. In combination with LC3 and/or TOMM20 immunostainings, it may offer a more integrative approach for the evaluation of autophagy and mitophagy under ER stress conditions. Such an approach may additionally deepen our understanding of the effects of the incretin mimetics in the degradative machinery and mitochondrial biogenesis. Last but not least, our present and previously published data [27] have implicated the STAT3 kinase in the signaling network through which the incretin mimetics switch neuronal fate towards survival under conditions of persistent ER stress. However, Akt network represents the canonical signaling pathway through which the incretins exert their neuroprotective effects in preclinical models and patients of neurodegenerative disorders [18, 46, 106]. Therefore, pharmacological and genetic manipulation of Akt and STAT3 kinases in neurons co-treated with thapsigargin and/or incretin mimetics offer an interesting avenue for future experimentation. The latter could be further combined with an experiment addressing how the cyclic-AMP pathway, lying downstream of incretin receptor stimulation, impacts pharmacological effects in UPR and mitochondrial function following persistent ER stress; previous work has demonstrated that the aforementioned pathway is essential for mitigating energy failure and mitochondrial dysfunction in Aβ-treated astrocytes [107], as well as for obscuring Chop in pancreatic β cells [108, 109]. Such an experimental setup would broaden further our current knowledge on which signaling network critically determines ER stress outcome and the mechanism of action of these peptides.
Conclusion
In conclusion, our study demonstrates the neuroprotective and restorative effects of Liraglutide and the novel dual GLP-1/GIP agonist upon persistent ER stress in human neurons, adding important information to our current knowledge of the biochemical mechanisms that underly these effects [18, 46]. It expands the range of effects that incretin signaling has on cell physiology processes in human neurons. Several clinical trials in AD or PD patients already have shown good protective effects, demonstrating that activating GLP-1 and GIP signaling is a valid target for drug development to treat these CNS diseases. As there is no disease-modifying drug treatment available for such diseases, it is worth focusing efforts on this drug target and to improve and enhance the protective drug effects to develop a viable treatment for PD that can modify disease progression.
FUNDING
We would like to acknowledge the Rosetrees Trust, UK and Alzheimer’s Research Society, UK, for financially supporting our research.
CONFLICT OF INTEREST
C.H. is a named inventor on several patent applications for the use of incretin mimetics as treatments for neurodegenerative disorders. The other authors declare no competing interests.
DATA AVAILABILITY
All data supporting the findings of this study are included in this published article and its supplementary information file. Raw data is available upon reasonable request.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JPD-220030.
REFERENCES
[1] | Obeso JA , Stamelou M , Goetz CG , Poewe W , Lang AE , Weintraub D , Burn D , Halliday GM , Bezard E , Przedborski S , Lehericy S , Brooks DJ , Rothwell JC , Hallett M , DeLong MR , Marras C , Tanner CM , Ross GW , Langston JW , Klein C , Bonifati V , Jankovic J , Lozano AM , Deuschl G , Bergman H , Tolosa E , Rodriguez-Violante M , Fahn S , Postuma RB , Berg D , Marek K , Standaert DG , Surmeier DJ , Olanow CW , Kordower JH , Calabresi P , Schapira AHV , Stoessl AJ ((2017) ) Past, present, and future of Parkinson’s disease: A special essay on the 200th Anniversary of the Shaking Palsy. Mov Disord 32: , 1264–1310. |
[2] | Costa CAD , Manaa WE , Duplan E , Checler F ((2020) ) The endoplasmic reticulum stress/unfolded protein response and their contributions to Parkinson’s disease physiopathology. Cells 9: , 2495. |
[3] | Mou Z , Yuan YH , Zhang Z , Song LK , Chen NH ((2020) ) Endoplasmic reticulum stress, an important factor in the development of Parkinson’s disease. Toxicol Lett 324: , 20–29. |
[4] | Kovacs G , Reimer L , Jensen PH ((2021) ) Endoplasmic reticulum-based calcium dysfunctions in synucleinopathies. Front Neurol 12: , 742625. |
[5] | Martinez G , Duran-Aniotz C , Cabral-Miranda F , Vivar JP , Hetz C ((2017) ) Endoplasmic reticulum proteostasis impairment in aging. Aging Cell 16: , 615–623. |
[6] | Martinez G , Khatiwada S , Costa-Mattioli M , Hetz C ((2018) ) ER proteostasis control of neuronal physiology and synaptic function. Trends Neurosci 41: , 610–624. |
[7] | Martinez-Vicente M , Vila M ((2013) ) Alpha-synuclein and protein degradation pathways in Parkinson’s disease: A pathological feed-back loop. Exp Neurol 247: , 308–313. |
[8] | Ramirez OA , Couve A ((2011) ) The endoplasmic reticulum and protein trafficking in dendrites and axons. Trends Cell Biol 21: , 219–227. |
[9] | Ktistakis NT ((2020) ) ER platforms mediating autophagosome generation. Biochim Biophys Acta Mol Cell Biol Lipids 1865: , 158433. |
[10] | Senft D , Ronai ZA ((2015) ) UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem Sci 40: , 141–148. |
[11] | Friedman JR , Lackner LL , West M , DiBenedetto JR , Nunnari J , Voeltz GK ((2011) ) ER tubules mark sites of mitochondrial division. Science 334: , 358–362. |
[12] | Korobova F , Ramabhadran V , Higgs HN ((2013) ) An actin-dependent step in mitochondrial fission mediated by the ER-associated formin INF2. Science 339: , 464–467. |
[13] | Schroder M , Kaufman RJ ((2005) ) The mammalian unfolded protein response. Annu Rev Biochem 74: , 739–789. |
[14] | Stutzmann GE , Mattson MP ((2011) ) Endoplasmic reticulum Ca(2+) handling in excitable cells in health and disease. Pharmacol Rev 63: , 700–727. |
[15] | Korkotian E , Segal M ((1998) ) Fast confocal imaging of calcium released from stores in dendritic spines. Eur J Neurosci 10: , 2076–2084. |
[16] | Mellstrom B , Naranjo JR ((2001) ) Mechanisms of Ca(2+)-dependent transcription. Curr Opin Neurobiol 11: , 312–319. |
[17] | Bravo R , Vicencio JM , Parra V , Troncoso R , Munoz JP , Bui M , Quiroga C , Rodriguez AE , Verdejo HE , Ferreira J , Iglewski M , Chiong M , Simmen T , Zorzano A , Hill JA , Rothermel BA , Szabadkai G , Lavandero S ((2011) ) Increased ER-mitochondrial coupling promotes mitochondrial respiration and bioenergetics during early phases of ER stress. J Cell Sci 124: , 2143–2152. |
[18] | Yang X , Feng P , Ji R , Ren Y , Wei W , Holscher C ((2022) ) Therapeutic application of GLP-1 and GIP receptor agonists in Parkinson’s disease. Expert Opin Ther Targets 26: , 445–460. |
[19] | Ferrari F , Moretti A , Villa RF ((2022) ) Incretin-based drugs as potential therapy for neurodegenerative diseases: Current status and perspectives. Pharmacol Ther 239: , 108277. |
[20] | Safar MM , Abdelkader NF , Ramadan E , Kortam MA , Mohamed AF ((2021) ) Novel mechanistic insights towards the repositioning of alogliptin in Parkinson’s disease. Life Sci 287: , 120132. |
[21] | Athauda D , Maclagan K , Skene SS , Bajwa-Joseph M , Letchford D , Chowdhury K , Hibbert S , Budnik N , Zampedri L , Dickson J , Li Y , Aviles-Olmos I , Warner TT , Limousin P , Lees AJ , Greig NH , Tebbs S , Foltynie T ((2017) ) Exenatide once weekly versus placebo in Parkinson’s disease: A randomised, double-blind, placebo-controlled trial. Lancet 390: , 1664–1675. |
[22] | Vijiaratnam N , Girges C , Auld G , Chau M , Maclagan K , King A , Skene S , Chowdhury K , Hibbert S , Morris H , Limousin P , Athauda D , Carroll CB , Hu MT , Silverdale M , Duncan GW , Chaudhuri R , Lo C , Del Din S , Yarnall AJ , Rochester L , Gibson R , Dickson J , Hunter R , Libri V , Foltynie T ((2021) ) Exenatide once weekly over 2 years as a potential disease-modifying treatment for Parkinson’s disease: Protocol for a multicentre, randomised, double blind, parallel group, placebo controlled, phase 3 trial: The ‘Exenatide-PD3’ study. BMJ Open 11: , e047993. |
[23] | Hogg E , Wu T , Bresee C , Wertheimer J , Malatt C , Tan E , Pomeroy H , Nuno M , Wyse R , Tagliati M ((2022) ) A phase II, randomized, double-blinded, placebo-controlled trial of liraglutide in Parkinson’s disease. SSRN https://papers.ssrn.com/sol3/papers.cfm?abstract_id=4212371 |
[24] | Edison P , Femminella G , Ritchie C , Holmes C , Walker Z , Ridha B , Raza S , Livingston N , Nowell J , Busza G , Frangou E , Love S , Williams G , Lawrence R , McFarlane B , Archer H , Coulthard E , Underwood B , Koranteng P , Karim S , Bannister C , Perneczky R , Prasanna A , Junaid K , McGuinness B , Nilforooshan R , Macharouthu A , Donaldson A , Thacker S , Russell G , Malik N , Mate V , Knight L , Kshemendran S , Hölscher C , Mansouri A , Chester-Jones M , Holmes J , Williams S , J Brooks D , Harrison J , Tadros G , Passmore A , Ballard C ((2021) ) Evaluation of liraglutide in the treatment of Alzheimer’s disease. Alzheimers Dement 17: , S9. |
[25] | Zhang ZQ , Holscher C ((2020) ) GIP has neuroprotective effects in Alzheimer and Parkinson’s disease models. Peptides 125: , 170184. |
[26] | Betzer C , Lassen LB , Olsen A , Kofoed RH , Reimer L , Gregersen E , Zheng J , Cali T , Gai WP , Chen T , Moeller A , Brini M , Fu Y , Halliday G , Brudek T , Aznar S , Pakkenberg B , Andersen JP , Jensen PH ((2018) ) Alpha-synuclein aggregates activate calcium pump SERCA leading to calcium dysregulation. EMBO Rep 19: , e44617. |
[27] | Panagaki T , Michael M , Holscher C ((2017) ) Liraglutide restores chronic ER stress, autophagy impairments and apoptotic signalling in SH-SY5Y cells. Sci Rep 7: , 16158. |
[28] | Panagaki T , Gengler S , Holscher C ((2018) ) The novel DA-CH3 dual incretin restores endoplasmic reticulum stress and autophagy impairments to attenuate Alzheimer-like pathology and cognitive decrements in the APPSWE/PS1DeltaE9 mouse model. J Alzheimers Dis 66: , 195–218. |
[29] | Thastrup O , Cullen PJ , Drobak BK , Hanley MR , Dawson AP ((1990) ) Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2(+)-ATPase. Proc Natl Acad Sci U S A 87: , 2466–2470. |
[30] | Panagaki T , Randi EB , Szabo C ((2020) ) Role of hydrogen sulfide and 3-mercaptopyruvate sulfurtransferase in the regulation of the endoplasmic reticulum stress response in hepatocytes. Biomolecules 10: , 653. |
[31] | Panagaki T , Randi EB , Szabo C ((2020) ) Role of 3-mercaptopyruvate sulfurtransferase in the regulation of proliferation and cellular bioenergetics in human Down syndrome fibroblasts. Biomolecules 10: , 1692. |
[32] | Perry SW , Norman JP , Barbieri J , Brown EB , Gelbard HA ((2011) ) Mitochondrial membrane potential probes and the proton gradient: A practical usage guide. Biotechniques 50: , 98–115. |
[33] | Wang Y , Shen J , Arenzana N , Tirasophon W , Kaufman RJ , Prywes R ((2000) ) Activation of ATF6 and an ATF6 DNA binding site by the endoplasmic reticulum stress response. J Biol Chem 275: , 27013–27020. |
[34] | Deniaud A , Sharaf el dein O , Maillier E , Poncet D , Kroemer G , Lemaire C , Brenner C ((2008) ) Endoplasmic reticulum stress induces calcium-dependent permeability transition, mitochondrial outer membrane permeabilization and apoptosis. Oncogene 27: , 285–299. |
[35] | Rossi A , Pizzo P , Filadi R ((2019) ) Calcium, mitochondria and cell metabolism: A functional triangle in bioenergetics. Biochim Biophys Acta Mol Cell Res 1866: , 1068–1078. |
[36] | Bhola PD , Letai A ((2016) ) Mitochondria-judges and executioners of cell death sentences. Mol Cell 61: , 695–704. |
[37] | Gimenez-Cassina A , Danial NN ((2015) ) Regulation of mitochondrial nutrient and energy metabolism by BCL-2 family proteins. Trends Endocrinol Metab 26: , 165–175. |
[38] | Ramsey CP , Glass CA , Montgomery MB , Lindl KA , Ritson GP , Chia LA , Hamilton RL , Chu CT , Jordan-Sciutto KL ((2007) ) Expression of Nrf2 in neurodegenerative diseases. J Neuropathol Exp Neurol 66: , 75–85. |
[39] | Reich N , Hölscher C ((2022) ) The neuroprotective effects of glucagon-like peptide 1 in Alzheimer’s and Parkinson’s disease: An in-depth review. Front Neurosci 16: , 970925. |
[40] | Cai Y , Arikkath J , Yang L , Guo ML , Periyasamy P , Buch S ((2016) ) Interplay of endoplasmic reticulum stress and autophagy in neurodegenerative disorders. Autophagy 12: , 225–244. |
[41] | Hetz C , Mollereau B ((2014) ) Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat Rev Neurosci 15: , 233–249. |
[42] | Ohno M ((2018) ) PERK as a hub of multiple pathogenic pathways leading to memory deficits and neurodegeneration in Alzheimer’s disease. Brain Res Bull 141: , 72–78. |
[43] | Santos LE , Ferreira ST ((2018) ) Crosstalk between endoplasmic reticulum stress and brain inflammation in Alzheimer’s disease. Neuropharmacology 136: , 350–360. |
[44] | Hetz C , Chevet E , Oakes SA ((2015) ) Proteostasis control by the unfolded protein response. Nat Cell Biol 17: , 829–838. |
[45] | Lin JH , Li H , Yasumura D , Cohen HR , Zhang C , Panning B , Shokat KM , Lavail MM , Walter P ((2007) ) IRE1 signaling affects cell fate during the unfolded protein response. Science 318: , 944–949. |
[46] | Hölscher C ((2022) ) Glucagon-like peptide 1 and glucose-dependent insulinotropic peptide hormones and novel receptor agonists protect synapses in Alzheimer’s and Parkinson’s diseases. Front Synaptic Neurosci 14: , 955258. |
[47] | Martinez G , Vidal RL , Mardones P , Serrano FG , Ardiles AO , Wirth C , Valdes P , Thielen P , Schneider BL , Kerr B , Valdes JL , Palacios AG , Inestrosa NC , Glimcher LH , Hetz C ((2016) ) Regulation of memory formation by the transcription factor XBP1. Cell Rep 14: , 1382–1394. |
[48] | Ogata M , Hino S , Saito A , Morikawa K , Kondo S , Kanemoto S , Murakami T , Taniguchi M , Tanii I , Yoshinaga K , Shiosaka S , Hammarback JA , Urano F , Imaizumi K ((2006) ) Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol 26: , 9220–9231. |
[49] | Liu D , Liu X , Zhou T , Yao W , Zhao J , Zheng Z , Jiang W , Wang F , Aikhionbare FO , Hill DL , Emmett N , Guo Z , Wang D , Yao X , Chen Y ((2016) ) IRE1-RACK1 axis orchestrates ER stress preconditioning-elicited cytoprotection from ischemia/reperfusion injury in liver. J Mol Cell Biol 8: , 144–156. |
[50] | Margariti A , Li H , Chen T , Martin D , Vizcay-Barrena G , Alam S , Karamariti E , Xiao Q , Zampetaki A , Zhang Z , Wang W , Jiang Z , Gao C , Ma B , Chen YG , Cockerill G , Hu Y , Xu Q , Zeng L ((2013) ) XBP1 mRNA splicing triggers an autophagic response in endothelial cells through BECLIN-1 transcriptional activation. J Biol Chem 288: , 859–872. |
[51] | Pickford F , Masliah E , Britschgi M , Lucin K , Narasimhan R , Jaeger PA , Small S , Spencer B , Rockenstein E , Levine B , Wyss-Coray T ((2008) ) The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest 118: , 2190–2199. |
[52] | Murphy KE , Gysbers AM , Abbott SK , Tayebi N , Kim WS , Sidransky E , Cooper A , Garner B , Halliday GM ((2014) ) Reduced glucocerebrosidase is associated with increased alpha-synuclein in sporadic Parkinson’s disease. Brain 137: , 834–848. |
[53] | Spencer B , Potkar R , Trejo M , Rockenstein E , Patrick C , Gindi R , Adame A , Wyss-Coray T , Masliah E ((2009) ) Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in alpha-synuclein models of Parkinson’s and Lewy body diseases. J Neurosci 29: , 13578–13588. |
[54] | Wang K , Huang J , Xie W , Huang L , Zhong C , Chen Z ((2016) ) Beclin1 and HMGB1 ameliorate the alpha-synuclein-mediated autophagy inhibition in PC12 cells. Diagn Pathol 11: , 15. |
[55] | Bulleid NJ , Ellgaard L ((2011) ) Multiple ways to make disulfides. Trends Biochem Sci 36: , 485–492. |
[56] | Pajares M , Cuadrado A , Rojo AI ((2017) ) Modulation of proteostasis by transcription factor NRF2 and impact in neurodegenerative diseases. Redox Biol 11: , 543–553. |
[57] | Bousette N , Abbasi C , Chis R , Gramolini AO ((2014) ) Calnexin silencing in mouse neonatal cardiomyocytes induces Ca2+ cycling defects, ER stress, and apoptosis. J Cell Physiol 229: , 374–383. |
[58] | Zanotto-Filho A , Masamsetti VP , Loranc E , Tonapi SS , Gorthi A , Bernard X , Goncalves RM , Moreira JC , Chen Y , Bishop AJ ((2016) ) Alkylating agent-induced NRF2 blocks endoplasmic reticulum stress-mediated apoptosis via control of glutathione pools and protein thiol homeostasis. Mol Cancer Ther 15: , 3000–3014. |
[59] | Simmen T , Lynes EM , Gesson K , Thomas G ((2010) ) Oxidative protein folding in the endoplasmic reticulum: Tight links to the mitochondria-associated membrane (MAM). Biochim Biophys Acta 1798: , 1465–1473. |
[60] | Kaufman RJ , Malhotra JD ((2014) ) Calcium trafficking integrates endoplasmic reticulum function with mitochondrial bioenergetics. Biochim Biophys Acta 1843: , 2233–2239. |
[61] | Nakagawa T , Yuan J ((2000) ) Cross-talk between two cysteine protease families. Activation of caspase-12 by calpain in apoptosis. J Cell Biol 150: , 887–894. |
[62] | Kitamura Y , Miyamura A , Takata K , Inden M , Tsuchiya D , Nakamura K , Taniguchi T ((2003) ) Possible involvement of both endoplasmic reticulum-and mitochondria-dependent pathways in thapsigargin-induced apoptosis in human neuroblastoma SH-SY5Y cells. J Pharmacol Sci 92: , 228–236. |
[63] | Morishima N , Nakanishi K , Takenouchi H , Shibata T , Yasuhiko Y ((2002) ) An endoplasmic reticulum stress-specific caspase cascade in apoptosis. Cytochrome c-independent activation of caspase-9 by caspase-12. J Biol Chem 277: , 34287–34294. |
[64] | Mungarro-Menchaca X , Ferrera P , Moran J , Arias C ((2002) ) beta-Amyloid peptide induces ultrastructural changes in synaptosomes and potentiates mitochondrial dysfunction in the presence of ryanodine. J Neurosci Res 68: , 89–96. |
[65] | Nakagawa T , Zhu H , Morishima N , Li E , Xu J , Yankner BA , Yuan J ((2000) ) Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature 403: , 98–103. |
[66] | Quiroz-Baez R , Ferrera P , Rosendo-Gutierrez R , Moran J , Bermudez-Rattoni F , Arias C ((2011) ) Caspase-12 activation is involved in amyloid-beta protein-induced synaptic toxicity. J Alzheimers Dis 26: , 467–476. |
[67] | B’Chir W , Chaveroux C , Carraro V , Averous J , Maurin AC , Jousse C , Muranishi Y , Parry L , Fafournoux P , Bruhat A ((2014) ) Dual role for CHOP in the crosstalk between autophagy and apoptosis to determine cell fate in response to amino acid deprivation. Cell Signal 26: , 1385–1391. |
[68] | B’Chir W , Maurin AC , Carraro V , Averous J , Jousse C , Muranishi Y , Parry L , Stepien G , Fafournoux P , Bruhat A ((2013) ) The eIF2alpha/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res 41: , 7683–7699. |
[69] | Komatsu M , Waguri S , Chiba T , Murata S , Iwata J , Tanida I , Ueno T , Koike M , Uchiyama Y , Kominami E , Tanaka K ((2006) ) Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441: , 880–884. |
[70] | Shiraishi D , Fujiwara Y , Komohara Y , Mizuta H , Takeya M ((2012) ) Glucagon-like peptide-1 (GLP-1) induces M2 polarization of human macrophages via STAT3 activation. Biochem Biophys Res Commun 425: , 304–308. |
[71] | Chiba T , Yamada M , Sasabe J , Terashita K , Shimoda M , Matsuoka M , Aiso S ((2009) ) Amyloid-beta causes memory impairment by disturbing the JAK2/STAT3 axis in hippocampal neurons. Mol Psychiatry 14: , 206–222. |
[72] | Chen HF , Liu SJ , Chen G ((2015) ) Heat shock protein 27 phosphorylation in the proliferation and apoptosis of human umbilical vein endothelial cells induced by high glucose through the phosphoinositide 3-kinase/Akt and extracellular signal-regulated kinase 1/2 pathways. Mol Med Rep 11: , 1504–1508. |
[73] | Park KW , Nozell SE , Benveniste EN ((2012) ) Protective role of STAT3 in NMDA and glutamate-induced neuronal death: Negative regulatory effect of SOCS3. PLoS One 7: , e50874. |
[74] | Walker CD , Long H , Williams S , Richard D ((2007) ) Long-lasting effects of elevated neonatal leptin on rat hippocampal function, synaptic proteins and NMDA receptor subunits. J Neurosci Res 85: , 816–828. |
[75] | Su Y , Zhang W , Patro CPK , Zhao J , Mu T , Ma Z , Xu J , Ban K , Yi C , Zhou Y ((2020) ) STAT3 regulates mouse neural progenitor proliferation and differentiation by promoting mitochondrial metabolism. Front Cell Dev Biol 8: , 362. |
[76] | MacPherson S , Horkoff M , Gravel C , Hoffmann T , Zuber J , Lum JJ ((2017) ) STAT3 regulation of citrate synthase is essential during the initiation of lymphocyte cell growth. Cell Rep 19: , 910–918. |
[77] | Demaria M , Giorgi C , Lebiedzinska M , Esposito G , D’Angeli L , Bartoli A , Gough DJ , Turkson J , Levy DE , Watson CJ , Wieckowski MR , Provero P , Pinton P , Poli V ((2010) ) A STAT3-mediated metabolic switch is involved in tumour transformation and STAT3 addiction. Aging (Albany NY) 2: , 823–842. |
[78] | Carlsson R , Ozen I , Barbariga M , Gaceb A , Roth M , Paul G ((2018) ) STAT3 precedes HIF1alpha transcriptional responses to oxygen and oxygen and glucose deprivation in human brain pericytes. PLoS One 13: , e0194146. |
[79] | Manning BD , Toker A ((2017) ) AKT/PKB signaling: Navigating the network. Cell 169: , 381–405. |
[80] | Xie X , Shu R , Yu C , Fu Z , Li Z ((2022) ) Mammalian AKT, the emerging roles on mitochondrial function in diseases. Aging Dis 13: , 157–174. |
[81] | Vander Haar E , Lee SI , Bandhakavi S , Griffin TJ , Kim DH ((2007) ) Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat Cell Biol 9: , 316–323. |
[82] | Saxton RA , Sabatini DM ((2017) ) mTOR signaling in growth, metabolism, and disease. Cell 169: , 361–371. |
[83] | Jung CH , Ro SH , Cao J , Otto NM , Kim DH ((2010) ) mTOR regulation of autophagy. FEBS Lett 584: , 1287–1295. |
[84] | Di Nardo A , Kramvis I , Cho N , Sadowski A , Meikle L , Kwiatkowski DJ , Sahin M ((2009) ) Tuberous sclerosis complex activity is required to control neuronal stress responses in an mTOR-dependent manner. J Neurosci 29: , 5926–5937. |
[85] | Heras-Sandoval D , Perez-Rojas JM , Hernandez-Damian J , Pedraza-Chaverri J ((2014) ) The role of PI3K/AKT/mTOR pathway in the modulation of autophagy and the clearance of protein aggregates in neurodegeneration. Cell Signal 26: , 2694–2701. |
[86] | Brewster JL , Linseman DA , Bouchard RJ , Loucks FA , Precht TA , Esch EA , Heidenreich KA ((2006) ) Endoplasmic reticulum stress and trophic factor withdrawal activate distinct signaling cascades that induce glycogen synthase kinase-3 beta and a caspase-9-dependent apoptosis in cerebellar granule neurons. Mol Cell Neurosci 32: , 242–253. |
[87] | Meares GP , Liu Y , Rajbhandari R , Qin H , Nozell SE , Mobley JA , Corbett JA , Benveniste EN ((2014) ) PERK-dependent activation of JAK1 and STAT3 contributes to endoplasmic reticulum stress-induced inflammation. Mol Cell Biol 34: , 3911–3925. |
[88] | Song L , De Sarno P , Jope RS ((2002) ) Central role of glycogen synthase kinase-3beta in endoplasmic reticulum stress-induced caspase-3 activation. J Biol Chem 277: , 44701–44708. |
[89] | Takadera T , Fujibayashi M , Kaniyu H , Sakota N , Ohyashiki T ((2007) ) Caspase-dependent apoptosis induced by thapsigargin was prevented by glycogen synthase kinase-3 inhibitors in cultured rat cortical neurons. Neurochem Res 32: , 1336–1342. |
[90] | Nijholt DA , Nolle A , van Haastert ES , Edelijn H , Toonen RF , Hoozemans JJ , Scheper W ((2013) ) Unfolded protein response activates glycogen synthase kinase-3 via selective lysosomal degradation. Neurobiol Aging 34: , 1759–1771. |
[91] | Hoozemans JJ , van Haastert ES , Nijholt DA , Rozemuller AJ , Eikelenboom P , Scheper W ((2009) ) The unfolded protein response is activated in pretangle neurons in Alzheimer’s disease hippocampus. Am J Pathol 174: , 1241–1251. |
[92] | Holmstrom KM , Kostov RV , Dinkova-Kostova AT ((2016) ) The multifaceted role of Nrf2 in mitochondrial function. Curr Opin Toxicol 1: , 80–91. |
[93] | Pajares M , Jimenez-Moreno N , Garcia-Yague AJ , Escoll M , de Ceballos ML , Van Leuven F , Rabano A , Yamamoto M , Rojo AI , Cuadrado A ((2016) ) Transcription factor NFE2L2/NRF2 is a regulator of macroautophagy genes. Autophagy 12: , 1902–1916. |
[94] | Zong ZH , Du ZX , Li N , Li C , Zhang Q , Liu BQ , Guan Y , Wang HQ ((2012) ) Implication of Nrf2 and ATF4 in differential induction of CHOP by proteasome inhibition in thyroid cancer cells. Biochim Biophys Acta 1823: , 1395–1404. |
[95] | Datta SR , Ranger AM , Lin MZ , Sturgill JF , Ma YC , Cowan CW , Dikkes P , Korsmeyer SJ , Greenberg ME ((2002) ) Survival factor-mediated BAD phosphorylation raises the mitochondrial threshold for apoptosis. Dev Cell 3: , 631–643. |
[96] | Chen H , Lin W , Zhang Y , Lin L , Chen J , Zeng Y , Zheng M , Zhuang Z , Du H , Chen R , Liu N ((2016) ) IL-10 promotes neurite outgrowth and synapse formation in cultured cortical neurons after the oxygen-glucose deprivation via JAK1/STAT3 pathway. Sci Rep 6: , 30459. |
[97] | Bakthisaran R , Tangirala R , Rao Ch M ((2015) ) Small heat shock proteins: Role in cellular functions and pathology. Biochim Biophys Acta 1854: , 291–319. |
[98] | Benn SC , Perrelet D , Kato AC , Scholz J , Decosterd I , Mannion RJ , Bakowska JC , Woolf CJ ((2002) ) Hsp27 upregulation and phosphorylation is required for injured sensory and motor neuron survival. Neuron 36: , 45–56. |
[99] | Stetler RA , Gao Y , Zhang L , Weng Z , Zhang F , Hu X , Wang S , Vosler P , Cao G , Sun D , Graham SH , Chen J ((2012) ) Phosphorylation of HSP27 by protein kinase D is essential for mediating neuroprotection against ischemic neuronal injury. J Neurosci 32: , 2667–2682. |
[100] | Xu Y , Diao Y , Qi S , Pan X , Wang Q , Xin Y , Cao X , Ruan J , Zhao Z , Luo L , Liu C , Yin Z ((2013) ) Phosphorylated Hsp27 activates ATM-dependent p53 signaling and mediates the resistance of MCF-7 cells to doxorubicin-induced apoptosis. Cell Signal 25: , 1176–1185. |
[101] | Mounir Z , Krishnamoorthy JL , Wang S , Papadopoulou B , Campbell S , Muller WJ , Hatzoglou M , Koromilas AE ((2011) ) Akt determines cell fate through inhibition of the PERK-eIF2alpha phosphorylation pathway. Sci Signal 4: , ra62. |
[102] | Gilman CP , Perry T , Furukawa K , Grieg NH , Egan JM , Mattson MP ((2003) ) Glucagon-like peptide 1 modulates calcium responses to glutamate and membrane depolarization in hippocampal neurons. J Neurochem 87: , 1137–1144. |
[103] | Perry T , Haughey NJ , Mattson MP , Egan JM , Greig NH ((2002) ) Protection and reversal of excitotoxic neuronal damage by glucagon-like peptide-1 and exendin-4. J Pharmacol Exp Ther 302: , 881–888. |
[104] | Sharma MK , Jalewa J , Hölscher C ((2014) ) Neuroprotective and anti-apoptotic effects of liraglutide on SH-SY5Y cells exposed to methylglyoxal stress. J Neurochem 128: , 459–471. |
[105] | Klionsky DJ , Abdel-Aziz AK , Abdelfatah S , Abdellatif M , Abdoli A , Abel S , Abeliovich H , Abildgaard MH , Abudu YP , Acevedo-Arozena A , et al. ((2021) ) Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy 17: , 1–382. |
[106] | Peng W , Zhou R , Sun ZF , Long JW , Gong YQ ((2022) ) Novel insights into the roles and mechanisms of GLP-1 receptor agonists against aging-related diseases. Aging Dis 13: , 468–490. |
[107] | Xie Y , Zheng J , Li S , Li H , Zhou Y , Zheng W , Zhang M , Liu L , Chen Z ((2021) ) GLP-1 improves the neuronal supportive ability of astrocytes in Alzheimer’s disease by regulating mitochondrial dysfunction via the cAMP/PKA pathway. Biochem Pharmacol 188: , 114578. |
[108] | Yusta B , Baggio LL , Estall JL , Koehler JA , Holland DP , Li H , Pipeleers D , Ling Z , Drucker DJ ((2006) ) GLP-1 receptor activation improves beta cell function and survival following induction of endoplasmic reticulum stress. Cell Metab 4: , 391–406. |
[109] | Rowlands J , Heng J , Newsholme P , Carlessi R ((2018) ) Pleiotropic effects of GLP-1 and analogs on cell signaling, metabolism, and function. Front Endocrinol (Lausanne) 9: , 672. |

