
Abstract
Despite sustained efforts to treat neurodegenerative diseases, little is known at the molecular level to understand and generate novel therapeutic approaches for these malignancies. Therefore, it is not surprising that neurogenerative diseases are among the leading causes of death in the aged population. Neurons require sophisticated cellular mechanisms to maintain proper protein homeostasis. These cells are generally sensitive to loss of gene expression control at the post-transcriptional level. Post-translational control responds to signals that can arise from intracellular processes or environmental factors that can be regulated through RNA-binding proteins. These proteins recognize RNA through one or more RNA-binding domains and form ribonucleoproteins that are critically involved in the regulation of post-transcriptional processes from splicing to the regulation of association of the translation machinery allowing a relatively rapid and precise modulation of the transcriptome. Neurotoxicity is the result of the biological, chemical, or physical interaction of agents with an adverse effect on the structure and function of the central nervous system. The disruption of the proper levels or function of RBPs in neurons and glial cells triggers neurotoxic events that are linked to neurodegenerative diseases such as spinal muscular atrophy (SMA), amyotrophic lateral sclerosis (ALS), fragile X syndrome (FXS), and frontotemporal dementia (FTD) among many others. The connection between RBPs and neurodegenerative diseases opens a new landscape for potentially novel therapeutic targets for the intervention of these neurodegenerative pathologies. In this contribution, a summary of the recent findings of the molecular mechanisms involved in the plausible role of RBPs in RNA processing in neurodegenerative disease is discussed.
Similar content being viewed by others


RNA-Binding Proteins: Overview
RNA-binding proteins (RBPs) can be defined as a wide group of proteins present in prokaryotic and eukaryotic cells that play a decisive role in regulating gene expression (Conti et al. 2017). Recently, RBPs have been described as the messenger ribonucleic acid (mRNA) clothes, as these proteins ensure that the different regions of mRNA (5′ and 3′ untranslated regions (UTR) and the coding region) could be either covered or exposed (Singh et al. 2015). These regulatory networks of RBP-mRNA-binding interactions, properly called ribonucleoprotein complexes (RNPs) (Thelen and Kye 2020), can remain stably bound throughout all post-transcriptional life of the mRNA (Lukong et al. 2008) and by these means allow numerous RNA interactions. Although these biochemical processes occur in numerous cells, these are especially essential in cells with complex RNA metabolisms, such as neurons and glial cells (Wolozin and Ivanov 2019).
The RNP association is extremely dynamic and prone to changes depending on the environment (Adeli 2011); any changes in some of the RNP could trigger cellular adaptive changes via modifications of the transcriptome and the proteome, meaning that RBPs are involved in the stabilization or decay of mRNAs in response to extracellular signals or stress (Alves 2016).
RBPs bind to the mRNA through a wide variety of intramolecular bonds in hairpins, stem loops, and other bumps and bulges (Attar 2014). Generally, RBPs associate with nascent mRNA both at the 5′- and 3′-ends. Their functions can generally be divided into nuclear and cytoplasmic activities. In the nucleus, RBPs regulate mRNA maturation, including RNA helicase activity, RNA polymerase elongation, splicing, and nuclear export. In the cytoplasm, RBPs regulate RNA transport, silencing, translation, and degradation (Halbeisen et al. 2008; Vanderweyde et al. 2013) (Fig. 1).

Representative summary of the different RBPs functions. The RBPs functions can be divided into nuclear and cytoplasmic activities. For example, in the nucleus, a) RBPs regulate the splicing of multi-exon genes and the exon skipping results in different protein isoforms from one unique gene. b) The RNA nuclear export by RBPs determines the proper out in the amount and correct timing from the nucleus. While in the cytoplasm, c) RBPs regulate mRNA stability and d) translation in the correct cytoplasmic localization
Despite the efforts to elucidate the number of RBPs expressed in eukaryotic cells, this number is still unknown; however, from studies based on bioinformatic strategies, it has been possible to calculate that between 2 and 8% of the number of total genes encode for RBPs (Gerstberger et al. 2014; Keene 2001).
It is important to note that not all RBPs share a common functional mechanism; it means that, while a subset of “housekeeping” RBPs could be constitutively and ubiquitously active, other subtypes are expressed in a more limited way such as those RBP involved in post-translational modifications (Xu et al. 2019), or being constantly inactive due to lack of their RNA targets (for example, due to the absence of RNA products from viral replication) (Garcia-Moreno et al. 2019).
Moreover, the regulatory roles of RBPs are also affected by the subcellular localization of RBPs and their RNA substrates (Nostrand et al. 2020). The transcripts are exported through nuclear pores to the cytoplasm in which RBPs may be targeted to specific subcellular regions by complexes consisting of motor proteins and RBPs or by a signal recognition particle (Halbeisen et al. 2008).
Modular Structures of RBP
The plethora interactions of RBPs and mRNA result from a high degree of modularity, most of them contain more than one RNA-binding domain that is arranged in different modules to meet their diverse functional requirements (Lunde et al. 2007). Most RNA-binding proteins are built from few RNA-binding units and possess sequences of 2 to 6 nucleotides capable of binding to RNA-binding domains (RBDs) (Burd and Dreyfusst 1994). Multiple copies allow the recognition of larger and more complex RNA targets; in addition, these modules endow a protein with the ability to bind RNA with higher specificity and affinity in comparison with individual domains (Shotwell et al. 2020; Maris et al. 2005; Zhou et al. 2014).
Functional Domains of RBP and their Properties
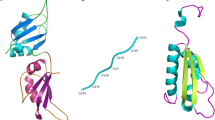
RBPs have one or more RNA-binding protein domains and share conserved domain structures and related functions (Vanderweyde et al. 2013); as shown in Fig. 2, most of these proteins fit the classical view of an RBP architecture with a modular combination of well-characterized RBDs and versatile RNA-binding surfaces (Beckmann et al. Jun 2016). The RBDs have been identified and classified according their substrate and structure: the RNA recognition motif (RRM), the zinc finger domain (ZnF), S1 domain, the K-homology domain (KH), the double-stranded RNA-binding domain (dsRBD), and Glycine-Arginine rich (RGG) (Conti et al. 2017; Chen and Varani 2005; Glisovic et al. 2008).

Modular structures of RBPs. Representative examples from some of the most common RNA-binding proteins involved in neurodegeneration. RNA-binding domains (RBDs) can act independently or when RBDs are found in multiple modules can act synergistically. Proteins are sized according to their amino acid lengths. RRM, RNA recognition motif; dsRBD, double-stranded RNA-binding motif; ZnF, zinc finger motif; KH, K-homology domain; RGG, Arg-Gly-Gly motiv; G, Gly motiv; Q/G/S/Y, Gln-Gly-Ser-Tyr motif. Modified from (Shotwell et al. 2020)
RNA Recognition Motif Domain
The RNA Recognition Motif (RRM) domain is an abundant domain and the most studied both in terms of structure and biochemistry. Genome sequencing studies have provided evidence that RRM-containing proteins are present in all forms of life (Maris et al. 2005; Afroz et al. 2015; Oliveira et al. 2017). To date, more than 10,000 RRMs have been identified that function practically in all post-transcriptional gene expression processes; in humans, ~ 0.5–1% of genes contain an RRM, often in multiple copies of the same polypeptide (Kuo et al. 2014).
Modified from Shotwell et al. (2020) The RRM is composed of a stretch of 80–90 amino acids that form a four-stranded antiparallel β sheet with two helices forming a divided αβ (βαββαβ) topology (Oubridge et al. 1994). The binding is mediated in most cases by an arginine (R) or a lysine (K) residue that forms a salt bridge to the phosphodiester backbone and two aromatic residues that make stacking interactions with nucleobases (Chen and Varani 2005). A single RRM can recognize a short sequence of nucleotides (between four and eight) due to the presence of exposed loops (Varani and Nagai 1998). Although some RNAs can bind to individual RRMs with high specificity, multiple domains are often required to define specificity because the number of nucleotides that are recognized by a single RRM is generally too small to define a single binding sequence (Duszczyk et al. 2019; Jankowsky and Harris 2015).
Other Binding Domains
The KH domain is approximately 70 amino acids long and binds four nucleotides. Two versions of the KH fold have been reported, type I and type II, which are found in eukaryotic and prokaryotic proteins, respectively (Valverde et al. 2008). The dsRBMs were first described as recognizing an RNA shape rather than an RNA sequence, and these domains contain approximately 70 amino acids and exhibit a conserved αβββα protein topology. These domains all interact along one face of a regular α-helix structure and can cover up to 16 bp spanning two consecutive minor grooves separated by a major groove (Chang and Ramos 2005; Stefl et al. 2005).
A classical ZnF is about 30 amino acids long and displays a ββα protein fold in which a β-hairpin and an α-helix are pinned together by a Zn2+ ion. In a single RBP, this motif can be found alone as a repeated domain or it can interact specifically with dsDNA motifs bases located in major grooves via side chains of residues present in their α-helix (Wolfe et al. 2000).
Low-complexity regions (LCR) are enriched mainly by serine (S), proline (P), glycine (G), arginine (R), lysine (K), and tyrosine (Y) (Toll-Riera et al. 2012). These amino acids form definite patterns: G often coexists with R or Y, generating repeats of RG or YG that can appear multiple times within a given protein region resulting in highly repetitive sequences. Both S and P have a high propensity to be in disordered regions (Schwartz et al. 2021). Many of the low-complexity regions in RBDs have been predicted to be intrinsically disordered regions (IDRs) that natively lack a three-dimensional stable structure (Calabretta and Richard 2015; Habchi et al. 2014; Uversky 2019).
RNA Processing and Regulation by RBPs
Crucial Role of RBP in mRNA Stability
After mRNA transcription in the nucleus, RBPs recognize pre-mRNA to regulate alternative splicing, polyadenylation, or stability. RPBs play an important role in mRNA stability; to make this possible, the RRM binds selectively to elements rich in adenylate and uridylate (ARE) in the 3′UTR region of mRNAs (Glisovic et al. 2008; Sena et al. 2021). AREs are present in 5–8% of human genes with various functions such as cell growth and differentiation, signal transduction, apoptosis, nutrient transport, and metabolism (García-Mauriño et al. 2017).
When the mature mRNA is transported to the cytoplasm, both its stability and distribution to different cell compartments can be modified by the different interactions with RBPs (Matoulkova et al. 2012). The final fate of the mRNA depends on the signaling pathway associated with the binding of the RBP in question; in general, RBPs can modify the half-life of the mRNA, that is, they can stabilize or destabilize it (Matoulkova et al. 2012). For example, proteins such as heterogeneous nuclear ribonucleoprotein D0 (hnRNPD0), tristetraprolin (TTP), TIA-1 T-cell internal antigen-1 (TIA-1), TIA-1 related protein (TIAR), and K-homology splicing regulatory protein (KSRP) bind to AREs and destabilize the mRNA, while the different proteins of the Hu family such as human antigen R (HuR) stabilize the mRNA as they delay the initiation of disintegration (García-Mauriño et al. 2017). Furthermore, with the help of RRM and the nuclear transport sequence (HNS), HuR binds to the target mRNAs in the nucleus, exports and protects it during cytoplasmic transit, and facilitates its recruitment into the translation machinery. This leads translation initiation increase the the mRNA stability (Fan and Steitz 1998). Interestingly, HuR proteins are susceptible to post-translational modifications. Their phosphorylation by protein kinase C (PKC) leads to increased translocation to the cytoplasm, resulting in altered cellular processes (Grammatikakis et al. 2017). Therefore, the presence of a nucleus or cytoplasmic HuR determines the normal or pathological state of a cell in the context of various physiological and pathological stimuli (Suresh et al. 2016).
RBPs, 3′UTRs, and poly (A) Tail of mRNA
Polyadenylation is an exquisite process that consists in the addition of a poly (A) tail to the mRNA by poly (A) polymerase; this phenomenon generates an effect on its nuclear transport, translation efficiency, and stability. All eukaryotic mRNAs, except the replication-dependent histone mRNAs, are polyadenylated at their 3′ ends in a process associated with transcriptional termination. It has been demonstrated in eggs and early embryos that mRNAS with longer poly (A) tails are more efficiently translated (Bartel and Xiang 2021; Moore and Lindern 2018). The binding of poly A-binding proteins (PABP) to the poly (A) tail is critical for the translation initiation. PABPs interact directly with the eukaryotic translation initiation factor 4G (eIF4G) scaffold protein of the eukaryotic initiation factor 4F (eIF4F) cap-binding complex (Brook and Gray 2012; Hinnebusch and Lorsch 2012). This brings the mRNA tail closer to the cap and forms an mRNA loop that optimizes the recycling of translation initiation and elongation factors (Neelagandan et al. 2020; Sonenberg and Hinnebusch 2009). Furthermore, the binding of PABP to the poly (A) tail protects from degradation, and the length of the poly (A) tail also affects the initiation of translation (Norbury 2013; Rissland et al. 2017).
The cytoplasmic polyadenylation element-binding protein (CPEB) is an RBP present in the cytoplasm responsible for the recruitment of poly (A) polymerase that has two RRM motifs and two ZnF (Hake et al. 1998; Ivshina et al. 2014; Kozlov et al. 2021), and by binding to the cytoplasmic polyadenylation element (CPE) 3′UTR, it recruits a series of proteins that interact to modulate the length of the tail of poly (A) resulting in positive regulation of translation (Richter 2007; Szostak and Gebauer 2013).
During development and in the adult organism, RBPs have key roles in the polyadenylation process. Alternative cleavage and polyadenylation (APA) lead to the expression of different isoforms of a same gene that might play a role in the etiology of a particular disease (Xing et al. Mar 2021). There are differential 3′UTRs lengths between cells in the organism that participates in the polyadenylation and cleavage regulation, for example, neurons are known to have longer 3′UTRs, while microglia and endothelial cells express shorter UTRs (Guvenek and Tian Sep 2018). The differential binding of RBPs at 3′UTR regulates the recruitment or prevents polyadenylation. Neuro-oncological ventral antigen (Nova) is another RBP that was found to bind 3′UTRs and enriched near poly (A) sites in the mouse brain. The comparison between Nova2 wild-type and knockout results in alternate 3′ UTR changes relative to total mRNA abundance, due to overlap with the canonical cleavage and polyadenylation specificity factor (CPSF) and/or cleavage stimulatory factor (CstF) binding sites of the Cugbp2 and Slc8a1 poly(A) sites, which are suppressed by Nova (Licatalosi et al. 2008). Muscleblind like splicing regulator 1 (MBNL) is another RBP essential polyadenylation regulator in mouse embryo fibroblasts; its depletion leads to dysregulation of thousands of alternative polyadenylation events (Batra et al. 2014).
RNA-binding protein fused in sarcoma (FUS) is frequently found around alternative polyadenylation (APA) sites of nascent RNA. APA sites located upstream of FUS cluster enhances polyadenylation by recruiting CPSF160 and up-regulates the alternative short transcript. In contrast, APA sites located downstream from FUS cluster polyadenylation is not activated; RNAP II-suppressing effect of FUS leads to down-regulation of the alternative short transcript. The regulation of mRNA lengths by FUS is operational in two-thirds of transcripts in neuronal cells, with enrichment in genes involved in synaptic activities (Masuda et al. 2015). In a structural and cis sense, the 3′UTR are key regions pre-mRNA regulated by RBPs for polyadenylation and splicing events.
RBP Association with Splicing Regulation
RBPs can regulate gene expression from the splice site through their binding to RNA (Yee et al. 2019). Constitutive splicing is the process of intron (non-coding segments) removal and exon (coding segments) ligation in the order in which they appear in a gene. Alternative splicing is a deviation from this preferred sequence resulting in various forms of mature mRNA (Wang et al. 2015). Most human intron deletions are catalyzed by a large RNP complex called a spliceosome. It is estimated that 95% of human genes are alternately spliced (Fredericks et al. 2015; Lee et al. 2015).
The two major RBPs splicing factors are the heterogeneous ribonucleoprotein particles (hnRNPs) and serine-arginine (SR) proteins (Fredericks et al. 2015). Although hnRNPs and SR proteins are believed to be the main splicing factors that regulate RBP, RNA-binding protein fused in sarcoma (FUS) has recently been implicated (Humphrey et al. 2020). Although there remains a class of uncharacterized hnRNP proteins, over 50% have been characterized to play a role in splicing (Han et al. 2013). SR proteins are distinguished due to their domain found near the C-terminal domain that promotes protein–protein interactions between the SR protein and the spliceosome (Zheng et al. 2020). Many RBPs regulate the skipping or inclusion exon according to downstream or upstream binding site, for example Nova 1 and 2, RNA-binding protein fox-1 (FOX-1), and polypyrimidine tract-binding protein (Ptbp) 1 and 2 (Ule et al. 2006; Raj and Blencowe 2015). Any change in the cis sequence and levels of trans-factors can alter splicing and cause disease.
Mechanisms of RBP-related Splicing Dysregulation
Pre-mRNA Mis-splicing
Removal of introns from pre-mRNAs is a sine-qua-non process for the expression of most human genes. Pre-mRNA splicing and its regulation require a complex array of cis-elements (the splicing code) embedded in pre-mRNAs and trans factors that bind to these elements (Chao et al. 2021).
Alternative splicing is influenced by interactions between RNA sequences and the surrounding sequence contexts: exonic splicing enhancer (ESE), exonic splicing silencer (ESS), intronic splicing enhancer (ISE), and intronic splicing silencer (ISS) elements and their binding transregulatory factors (e.g., splicing factors and RBPs) (Chao et al. 2021; Singh and Cooper 2012). RNA recognition motifs interact with single-stranded RNA targets using their β-sheet surface. Diseases caused by point mutations can alter splicing by the disruption of cis-elements that modulate the recognition of the splice sites. These auxiliary elements are often ligands for RBPs (Fredericks et al. 2015). The main RBPs involved in alternative splicing are SR and hnRNP. SR proteins bound in the exon are generally regarded as activating splicing whereas the same protein at the intron can act as a repressor. Conversely, hnRNPs are regarded as repressors when bound to exonic locations and activators when bound to the intron (Erkelenz et al. 2013). ESE motifs functionally repress splicing when are found in the intron, becoming intronic splicing silencers (Martinez-Contreras et al. 2006). Likewise, ESS motifs have been shown to function as intronic splicing enhancers (Kanopka et al. 1996). Mutations that increase the stability of interactions between an RNA species and RBP substrate can cause disease. This has been demonstrated in several well-studied diseases particularly neurological and muscular degenerative disorders. Often, the repeated sequence becomes pathogenic after expanding beyond a threshold length. The toxic mRNA transcripts produced cause the dysregulation of alternative splicing of many pre-mRNAs in trans simultaneously, also known as spliceopathy (Afroz et al. 2015; Boo and Kim 2020).
Splicing Factors Alteration
Another category of spliceopathy is the direct mutation of a splicing factor. NOVA and TAR DNA-binding protein 43 (TDP-43) are the two RBPs in this category of spliceopathy, regulate alternative events in neurons, and the loss of either of them results in severe pathogenesis (Boo and Kim 2020; Kapeli et al. 2017). Loss of Nova proteins, as a result of an autoimmune paraneoplastic neurological disorder, manifests itself in neurological symptoms of excess motor movements (paraneoplastic myoclonic opsoclonus ataxia, POMA), on other hand, TPD-43 has been involved in ALS pathology (Buckanovich et al. 1993).
RBP are Involved in Stress Granules
In eukaryotic cells, stress conditions such as accumulation of reactive oxygen species (ROS) often inhibit the initiation of translation and trigger the formation of cytoplasmic RNA–protein complexes called stress granules (SG) (Markmiller et al. 2018) that store RNA and that is released when physiological conditions are restored (Shashidharan et al. 1997; Schieweck et al. 2021). These granules are present in the pathophysiology of neurodegenerative diseases, particularly amyotrophic lateral sclerosis, frontotemporal dementia, and Alzheimer’s disease (AD). SGs contain 40S ribosomal subunits, mRNA, translation initiation factors, and RBPs (Wolozin and Ivanov 2019; Markmiller et al. 2018) so that post-translational modifications of RBP can directly modify the regulation of SGs (Anderson and Kedersha 2009). Recent studies show that few nuclear RBPs are expressed under neuropathological conditions, and a higher fraction is present in SG (Apicco et al. 2018; Armstrong et al. 2012; Janssens et al. 2013). Through proteomics, it has been demonstrated that the following factors contribute to the assembly of SG:
Interaction with the G3BP stress granule assembly factor 1 (G3BP1) protein: RNAs containing G-quadruplex (GGGGCC) act as molecular scaffolds to recruit specific RBPs such as G3BP1, increasing their local concentrations and promoting SG (Hofmann et al. 2020; Fay et al. 2017; Jain et al. 2016).
The dynamic assembly of SG is also promoted by RBPs such as TIA-1 and TIAR since these proteins are capable of dimerizing and promoting polysome assembly (Fig. 3) (Martin and Tazi 2014) in response to phosphorylation of the eukaryotic initiation factor 2 alpha (eIF-2α). TIA-1 and TIAR have 3 RRMs which confer the necessary functions for the assembly of SG (Kedersha et al. 2000; Panas et al. 2016).
SG assembly can also be affected by changes in levels of ubiquitination (Cao et al. 2020) or methylation (Xie and Denman 2011). Stress granules also interact with P-bodies (PB), another dynamic cytoplasmic RNA protein structure found in the cytoplasm of eukaryotes. PB contains components of the mRNA decomposition machinery (Buchan and Parker 2009).
SG and PB interact physically, suggesting possible traffic of stored mRNA between these compartments (Parker and Sheth 2007). PB recruits mRNA for translation control (Hubstenberger et al. 2017) and degradation (Fig. 4) (Decker and Parker 2012).

Assembly and disassembly of stress granules. Under unstressed conditions, mRNA exists in the cytoplasm and is normally translated. Upon stress, mRNAs are protected within stress granules. Once the stress has been removed, the stress granules disassemble. The dynamic assembly of SG is also promoted by RBPs such as TIA-1. TDP-43, TAR DNA-binding protein 43; TIA-1, T-cell internal antigen-1; G3BP1, stress granule assembly factor 1; APEX2, apurinic/apyrimidinic endodeoxyribonuclease 2. Modified from (Hofmann et al. 2020)

RNA regulation cascade by RBPs from the nucleus to the cytoplasm. From DNA to RNA transcription, there are RBPs involved in the isoform length by two mechanisms: the alternative exons selection (splicing) and the alternative polyadenylation sites. After RNA export at the post-translational level is regulated by some RBPs that can conduce to decoy, protection mechanisms such as the formation of dynamic stress granules and P-bodies or ensure the performance of mRNA through translating polysomes that ensure a high peptide-protein expression rate, all processes in at post-translational level
SG Disassembly
SGs contain non-translatable mRNAs that are classified and processed according to metabolic status and environmental changes, and these mRNAs can be activated when stressful conditions dissipate repression or decomposition. Theoretically, SG could be disassembled by dissociating the interactions by transferring the material to a PB or removing the mRNAs from SGs by entering the polysomes (Markmiller et al. 2018; Anderson et al. 2015).
RBP are Essential for Brain Function
To ensure a proper neuronal development and synaptic plasticity, the brain contains the highest amount of transcriptional and post-transcriptional mechanisms described thus far (Pilaz and Silver 2015). Following RNA splicing and the first step of quality control in the nucleus, mRNAs will be exported through the nuclear pore into the cytoplasm, where the interaction with different RBPs ensures proper control of localization, stability, and translation (Darnell and Richter 2012; Soheilypour and Mofrad 2018). The neuronal transcriptome is enormously diverse due to alternative splicing, polyadenylation, intron retention, and the occurrence of non-canonical coding sequences (Sibley et al. 2016). To guide the expression of the transcriptome, multiple RBPs will dynamically interact in a spatially and temporally defined as well as cell type-specific manner which explains the great variety of RBPs in cells (Schieweck et al. 2021). RBPs bound to mRNAs and are associated with motor proteins to specific localizations in membrane-less and shape high molecular weight complexes (mRNP). The kind of RBP determines the subcellular location of their components including mRNAs. In the brain, the axonal cone growth has a differential translational where the RBPs CPEB1 (Richter 2007) and ZBP1 (Huttelmaier et al. 2005) are critical mediators of mRNA transport and its translation. ZBP1 assembly β-actin mRNA to direct localization in axons and dendrites (Song et al. 2015). Staufen is another RBP involved with RNA granules moving along microtubules into dendrites of hippocampal neurons in a bidirectional manner (Kohrmann et al. 1999). The fragile X mental retardation protein (FMRP) interacts with kinesin to dendritic mRNA localization and regulates the local translation in these sites (Wang et al. 2016; Dictenberg et al. 2008). Pumilio 2 (Pum2) acts like FMRP as a translational regulator, and its specific localization is related to repressor function by inhibiting translation and promoting mRNA decay (Goldstrohm et al. 2018).
RBPs Dysregulation: Triggering the Disease
From the progress in genetic studies, it has been established that RBP dysregulation or mutation can trigger loss of neurons, neuronal function, and neurodegeneration (Kapeli et al. 2017). Nowadays, more and more RBPs are being recognized as causal factors or associated with neurological diseases, autoimmunity, and cancer (Wolozin and Ivanov 2019; Van Nostrand et al. 2020) reinforcing the importance of RBPs in the maintenance of the normal physiology of the CNS. A considerable amount of RBPs have LCR, so these proteins are prone to structural modifications and consequently trigger the loss or gain of function, contributing to the severity of neurodegeneration (Kapeli et al. 2017). Despite the complexities that RBPs currently represent, the binding sites in RNAs could provide more detailed information on the development of neuronal diseases that involve RBPs, which would undoubtedly be an important contribution to this public health problem (Pan et al. 2020).
Mutations in genes encoding RBP have been observed in patients with motor neuron disorders such as amyotrophic lateral sclerosis (ALS), spinal muscular atrophy (SMA), multisystem proteinopathy (MSP), and frontotemporal degeneration (FTD); of all these, ALS is the most common motor neuron disorder in adults; this condition is characterized by the progressive loss of motor neurons triggering fatal paralysis (Robberecht and Eykens 2015).
In neurons, mRNAs can be transported to and from axons and dendrites as mRNA can be translated locally. In neuronal processes, such as dendrites, there are a great variety of mRNAs and a large part of the transcriptome is present both in dendrites and in axons. In the vertebrate brain, mRNAs containing localization elements or “zip codes” have been identified in neuronal processes, including those encoding structural proteins (Tiruchinapalli et al. 2003), receptors (Grooms et al. 2006), and signaling molecules (Martin and Ephrussi 2009; Terenzio et al. 1421). These cis-acting sequences located in the 3′UTR are generally recognized by RBPs and will assemble into an RNP (Turner-Bridger et al. 2020). The cis information contained in the 3′UTR mRNAs is essential to the RBPs recognition; the activated leukocyte cell adhesion molecule (ALCAM) mediates homophilic adhesion of axons from the same neuronal subtype and is required for the formation of axon bundles. The lack of its 3′UTR results in overexpression and induces axon bundle aggregation and prevents axonal growth, whereas a decreased expression results in fasciculation (Thelen et al. 2012), accordingly. The full length of ALCAM maintains the right amount.
The RNP complex can mediate interactions with the translation machinery and self-assemble into transport granules (Eliscovich and Singer 2017). One of the most abundant RNPs in both axons and dendrites is β-actin, and its association with the zip code binding protein 1 (ZBP1) is essential for transport and proper localization (Biswas et al. 2019; Das and Yoon 2019).
Late endosomes serve as a platform for local axonal translation by binding to RBP, ribosomes, and mRNA (Cioni et al. 2019). Another interesting cellular component that regulates neuronal protein synthesis are RNA granules. Neurons contain various RNA granules, including SG and PB (Thelen and Kye 2020). Importantly, it has been shown that RNP granules located in dendrites can disassemble and mRNAs can be used as a template to produce synaptic proteins (Schieweck et al. 2021; Krichevsky and Kosik 2001). This process is crucial for neuronal health and function, as it is a cellular homeostatic mechanism for managing external stress and controlling synaptic plasticity. These mechanisms will directly impact the composition of the local proteome (Das and Yoon 2019; Jung et al. 2014).
As RBPs determine the axonal or dendritic mRNA repertoire as well as proteomes by trafficking mRNAs and regulating local protein synthesis, RBP plays a crucial role in neuronal function. Dysfunctional RNA processing in neuronal tissue plays a crucial role in neuronal pathology and is often observed in neurodegenerative diseases (Thelen and Kye 2020). Moreover, RNP plays an important role in RNA metabolism, regulating ribosome formation, spliceosomes, and silencing complexes. When gene mutations or deletions occur in neurons or a well-misregulated assembly of RNP occurs, it results in neuron degeneration which can lead to SMA, ALS, fragile X syndrome (FXS), among other pathologies (Shukla and Parker 2016).
Due to complex neuronal compartmentalization, it is indispensable to provide local mRNA transcripts and make neurons vulnerable to any change and loss of RNPs complex. Neurons can synthesize proteins at the synaptic compartment in response to many stimuli. For example, the cellular components necessary to produce proteins such as ribosomes and mRNAs are detected at the synaptic area (Ainsley et al. 2014; Poulopoulos et al. 2019). Thus, RNPs deliver specific sets of mRNAs and produce different proteins in particular subcellular compartments, due to the motifs that provide an accurate function of RBP and RNPs at local mRNA translation. These processes are crucial for neuronal development and function (Jung et al. 2014).
Role of RBPs in Neurotoxicity
The brain is susceptible to damage by several toxic agents such as metals, microorganisms, persistent organic pollutants, and high levels of glutamate. RBPs as key regulators of transcriptome are found deregulated in many neurotoxic diseases. In FTD and ALS, there are abnormal controls of mRNA translation by TDP-43 and FUS accumulation in SGs. The motor neuron degeneration in these diseases is related to mutations in RBPs genes (Gowell et al. 2021; Zhou et al. 2020; Vance et al. 2009).
The abnormal cytoplasmic accumulation of TDP-43, correctly called TDP-43 proteinopathy, contributes to neurotoxicity and the oligonucleotides treatment composed of TDP-43 target sequences rescues neurotoxicity (Schieweck et al. 2021; Mann et al. 2019). ALS-associated mutations in TDP-43 are frequently found in LCR Gly-rich domains that regulate phosphorylation and ubiquitination sites (Pesiridis et al. 2009). The prion-related domains rich in glutamine(Q) and asparagine (N) present in TDP-43, TIA-1, and FUS are associated with a highly prone to aggregation (Udan and Baloh 2011). As described above, the cytosolic accumulation of almost any RBPs and the disruption of their nuclear functions is a triggering feature of neurotoxicity. For example, wild-type human TDP-43 can be toxic when expressed in a heterologous C. elegans system or overexpressed in a cell culture model (Ash et al. 2010). In mice, TDP-43 mutant alleles cause dose-dependent asymmetrical motor axon withdrawal and the lethality and cognitive dysfunction are rescued with functional TDP-43 (Ebstein et al. 2019). The wild-type human TDP-43 expression causes mitochondrial aggregation, motor deficits, and early mortality in transgenic mice (Xu et al. 2010). In chick embryo models, TDP-43Q331K and TDP-43M337V showed a dramatic reduction in maturation compared to TDP-43WT with a failure to develop normal limbs and tail buds (Sreedharan et al. 2008). The lacking TDP-43 in flies results in deficient locomotive behaviors, life span reduction, and anatomical defects at the neuromuscular junctions (Feiguin et al. 2009). Some studies report that TDP-43 mutations are more neurotoxic compared to wild-type TDP-43; however, it is necessary to emphasize that a mutation in this RBP is not necessary to promote ALS (Gregory et al. 2020; Wegorzewska et al. 2009). Although some studies report neurodegeneration in the absence of cytosolic aggregation how consequence from TDP-43 specific localization to motor neuron nuclei (Hanson et al. 2010).
From RNA interference screening, the inositol-1,4,5-triphosphate receptor type 1 (ITPR1, mediator of Ca2+ efflux) was identified as a new regulator of nucleocytoplasmic transport of TDP-43 since the silencing of this receptor promotes the cytosolic accumulation of TDP-43. Therefore, these findings also suggest that the expression and localization of TDP-43 are regulated by Ca2+ (Kim et al. 2012). Duan et al. (2019) revealed that PARylation levels are an important regulator of assembly and disassembly dynamics of RNP granules containing hnRNP A1 and TDP-43. They also showed that both genetic and pharmacological inhibition of PARP mitigates neurotoxicity mediated by hnRNP A1 and TDP-43 in cellular and Drosophila models of ALS. At the same time, PAR binding through the hnRNP A1 PAR-binding motif regulates its association with stress granules (Duan et al. 2019).
Mutations in Matrin 3 (MATR3), a DNA and RNA-binding protein little studied so far, have also been described as causing ALS and FTD. Using a primary neuron model to evaluate MATR3-mediated neurotoxicity, Malik et al. (2018) showed that neurons were bidirectionally vulnerable to MATR3 levels. In addition, the ZnF MATR3 domains partially modulated toxicity; however, the elimination of their motifs for RNA recognition did not affect neuronal survival. On the other hand, contrary to other RBPs related to ALS, the cytoplasmic redistribution of MATR3 mitigated neurodegeneration, suggesting that nuclear MATR3 mediates toxicity (Malik et al. 2018).
Another of the main cause of ALS and FTD is the expanded GGGGCC (G4C2)n repeats in the first intron of the C9orf72 gene; this repetition promotes a gain of function that undoubtedly alters the homeostasis of post-transcriptional processes. Celona et al. (2017) identified Zfp106, a ZnF domain RBP, as a specific 4G RNA repeat binding protein. Furthermore, the authors showed that Zfp106 interacts with other RBPs. Zfp106 potently suppresses neurotoxicity in a Drosophila model of ALS C9orf72 (Celona et al. 2017). Another RBP in these diseases is the RNA editing enzyme adenosine deaminase acting on RNA 2 (ADAR2), which is mislocalized in C9orf72 repeat expansion = mediated ALS/FTD. Because of this mislocalization, severe RNA editing alterations were observed in multiple brain regions. The mislocalization of ADAR2 in C9orf72-mediated ALS/FTD is responsible for the alteration of RNA processing events that may impact vast cellular functions, including the integrated stress response (ISR) and protein translation (Moore et al. 2019).
In autism disorder (ASD), CPEB4 regulates the translation of specific mRNAs by modulating their poly(A)-tails, and it was found to bind transcripts of most high-confidence ASD risk genes. Individuals with idiopathic ASD show imbalances in CPEB4 transcript isoforms, and 9% of the transcriptome shows reduced poly(A)-tail length (Parras et al. 2018). In the same disease, functional defects of the cerebral cortex contribute to the clinical symptoms of ASD, and impairment of Rbfox1-iso1 is a main effector. The Rbfox1-iso1 knockdown in hippocampal neurons resulted in the reduction of primary axon length, total length of dendrites, spine density, and mature spine number with an important impact on neuronal migration and synapse network formation during corticogenesis (Hamada et al. 2016).
Taken together, the literature supports that RBPs are key regulators in many neurotoxicology diseases; in Table 1, we summarized the association of different RBPs dysfunction and process altered in neurological disorders.
Concluding Remarks
In this contribution, we have summarized our current knowledge on the role of RBPs in neurotoxicity. Numerous RBPs are involved in different stages of post-transcriptional control such as alternative splicing, SG dynamics, and mRNA localization. A dysregulation of RBPs to cell stress response at any level may be harmful to neuronal integrity and neuroplasticity. It has been experimentally proven that RBP disorders participate in different pathologies of the CNS and that the main diseases associated with TDP-43 proteinopathy are visualized in motor neuron disorders such as FTD and ALS. The hallmark of RBPs may help elucidate a new perspective involved in neurotoxicity mechanisms. Although some molecular mechanisms associated with RBP functions have been characterized, these are far from being fully understood, elucidating the pathogenesis associated with dysfunctional RBPs and altered local translation could contribute to discovering new drugs that could alleviate the pathology of neurological diseases.
Abbreviations
- 4G:
-
GGGGCC repeats
- AD:
-
Alzheimer disease
- ADAR2:
-
Adenosine deaminase acting on RNA 2
- ALCAM:
-
Activated leukocyte cell adhesion molecule
- ALS:
-
Amyotrophic lateral sclerosis
- APA sites:
-
Alternative polyadenylation sites
- APEX2:
-
Apurinic/apyrimidinic endodeoxyribonuclease 2
- ARE:
-
Adenylate and uridylate rich elements
- ASD:
-
Autism disorder
- ASD:
-
Autism spectrum disorder
- CEPB :
-
Cytoplasmic polyadenylation element-binding protein
- CNS:
-
Central nervous system
- COPD:
-
Chronic obstructive pulmonary disease
- CPE:
-
Cytoplasmic polyadenylation element
- CPSF:
-
Cleavage and polyadenylation specificity factor
- CstF:
-
Cleavage stimulatory factor
- Cugbp2 :
-
Elav-like family member 2
- dsRBD:
-
Double-stranded RNA-binding motif
- eIF4F:
-
Eukaryotic initiation factor 4F
- eIF4G:
-
Eukaryotic translation initiation factor 4G
- ESE:
-
Exonic splicing enhancer
- ESS:
-
Exonic splicing silencer
- FMRP:
-
Fragile X mental retardation protein
- FOX-1:
-
RNA-binding protein Fox-1 Homolog 1
- FTD:
-
Frontotemporal lobar dementia
- FUS:
-
RNA-binding protein fused in sarcoma
- FXS:
-
Fragile X syndrome
- G rich:
-
Glycine rich motif
- G3BP1:
-
G3BP stress granule assembly factor 1
- hnRNPD0:
-
Heterogeneous nuclear ribonucleoprotein D0
- hnRNPs:
-
Heterogeneous ribonucleoprotein particles
- HNS:
-
Nuclear transport sequence
- HuR:
-
Human antigen R
- IDR:
-
Intrinsically disordered regions
- IPF:
-
Idiopathic pulmonary fibrosis
- ISE:
-
Intronic splicing enhancer
- ISR:
-
Integrated stress response
- ISS:
-
Intronic splicing silencer
- ITPR1:
-
Inositol-1,4,5-triphosphate receptor type 1
- K rich:
-
Lysine rich motif
- KH:
-
K-homology domain
- KSRP:
-
K-homology splicing regulatory protein
- LCR:
-
Low-complexity regions
- MATR3:
-
Matrin 3
- MBNL:
-
Muscleblind like splicing regulator 1
- MD:
-
Myotonic dystrophy
- mRNA:
-
Messenger ribonucleic acid
- MSP:
-
Multisystem proteinopathy
- Nova:
-
Neuro-oncological ventral antigen
- P rich:
-
Proline-rich motif
- PABP:
-
Binding of Poly A binding proteins
- PARP:
-
Poly ADP-ribose polymerase
- PB:
-
P-bodies
- PEM:
-
Paraneoplastic encephalomyelitis
- PKC:
-
Protein kinase C
- POMA:
-
Paraneoplastic myoclonic opsoclonus ataxia
- Ptbp1/2:
-
Polypyrimidine tract-binding proteins 1/2
- Q/G/S/Y:
-
Gln-Gly-Ser-Tyr motif
- R rich:
-
Arginine rich motif
- RBD:
-
RNA-binding domains
- RBP:
-
RNA-binding proteins
- RGG:
-
Arg-Gly-Gly motif
- RNA:
-
Ribonucleic acid
- RNP:
-
Ribonucleoprotein
- ROS:
-
Reactive oxygen species
- RRM:
-
RNA recognition motif
- S rich:
-
Serine rich motif
- SCA2:
-
Spinocerebral ataxia type 2
- SG:
-
Stress granules
- Slc8a1 :
-
Solute carrier family 8 member A1
- SMA:
-
Spinal muscular atrophy
- SR:
-
Serine-arginine proteins
- SSN:
-
Subacute sensory neuropathy
- TDP-43:
-
TAR DNA-binding protein 43
- TIA-1:
-
T-cell internal antigen-1
- TIAR:
-
TIA-1 related protein
- TTP:
-
Tristetraprolin
- UTR:
-
Untranslated regions
- Y rich:
-
Tyrosine rich motif
- ZBP1:
-
Zip code binding protein 1
- ZnF:
-
Zinc finger motif
References
Adeli K (2011) Translational control mechanisms in metabolic regulation: critical role of RNA binding proteins, microRNAs, and cytoplasmic RNA granules. Am J Physiol Endocrinol Metabol 301 (6). https://doi.org/10.1152/ajpendo.00399.2011
Afroz T, Cienikova Z, Cléry A, Allain FHT (2015) One, two, three, four! How multiple RRMs read the genome sequence. Methods Enzymol 558(1):235–278. https://doi.org/10.1016/bs.mie.2015.01.015
Ainsley JA, Drane L, Jacobs J, Kittelberger KA, Reijmers LG (2014) Functionally diverse dendritic mRNAs rapidly associate with ribosomes following a novel experience,” (in eng). Nat Commun 5:4510
Alves LR (2016) RNA-binding proteins related to stress response and differentiation in protozoa. World J Biol Chem 7(1):78–78. https://doi.org/10.4331/wjbc.v7.i1.78
Anderson P, Kedersha N (2009) RNA granules: post-transcriptional and epigenetic modulators of gene expression. Nat Rev Mol Cell Biol 10(6):430–436. https://doi.org/10.1038/nrm2694
Anderson P, Kedersha N, Ivanov P (2015) Stress granules, P-bodies and cancer. Biochimica Et Biophysica Acta - Gene Regulatory Mechanisms 1849(7):861–870. https://doi.org/10.1016/j.bbagrm.2014.11.009
Apicco DJ et al (2018) Reducing the RNA binding protein TIA1 protects against tau-mediated neurodegeneration in vivo. Nat Neurosci 21(1):72–82. https://doi.org/10.1038/s41593-017-0022-z
Armstrong RA, Carter D, Cairns NJ (2012) A quantitative study of the neuropathology of 32 sporadic and familial cases of frontotemporal lobar degeneration with TDP-43 proteinopathy (FTLD-TDP). Neuropathol Appl Neurobiol 38(1):25–38. https://doi.org/10.1111/j.1365-2990.2011.01188.x
Ash PEA et al (2010) Neurotoxic effects of TDP-43 overexpression in C. elegans. Hum Mol Genet 19(16):3206–3218. https://doi.org/10.1093/hmg/ddq230
Attar N (2014) The RBPome: Where the brains meet the brawn. Genome Biol 15(1):1–5. https://doi.org/10.1186/gb4153
Bampton A, Gittings LM, Fratta P, Lashley T, Gatt A (2020) The role of hnRNPs in frontotemporal dementia and amyotrophic lateral sclerosis. Acta Neuropathol 140(5):599–623. https://doi.org/10.1007/s00401-020-02203-0
Banerjee A, Apponi LH, Pavlath GK, Corbett AH (2013) PABPN1: Molecular function and muscle disease. FEBS J 280(17):4230–4250. https://doi.org/10.1111/febs.12294
Barraud P, Allain FH (2012) “ADAR proteins: double-stranded RNA and Z-DNA binding domains”, (in eng). Curr Top Microbiol Immunol 353:35–60. https://doi.org/10.1007/82_2011_145
Bartel DP, Xiang K (2021) The molecular basis of coupling between poly(A)-tail length and translational efficiency. bioRxiv 2021.01.18.427055
Batra R et al (2014) Loss of MBNL leads to disruption of developmentally regulated alternative polyadenylation in RNA-mediated disease. Mol Cell 56(2):311–322. https://doi.org/10.1016/j.molcel.2014.08.027
Beckmann BM, Castello A, Medenbach J (2016) The expanding universe of ribonucleoproteins: of novel RNA-binding proteins and unconventional interactions. Pflugers Arch 468(6):1029–1040. https://doi.org/10.1007/s00424-016-1819-4
Biswas J, Nunes L, Das S, Yoon YJ, Eliscovich C, Singer RH (2019) Zipcode binding protein 1 (ZBP1; IGF2BP1): a model for sequence specific RNA regulation. Cold Spring Harb Symp Quant Biol 84:1–10. https://doi.org/10.1101/sqb.2019.84.039396.Zipcode
Bondy-Chorney E et al (2016) Staufen1 regulates multiple alternative splicing events either positively or negatively in DM1 indicating its role as a disease modifier. PLoS Genet 12(1):1–22. https://doi.org/10.1371/journal.pgen.1005827
Boo SH, Kim YK (2020) The emerging role of RNA modifications in the regulation of mRNA stability. Exp Mol Med 52(3):400–408. https://doi.org/10.1038/s12276-020-0407-z
Brook M, Gray NK (2012) The role of mammalian poly(A)-binding proteins in co-ordinating mRNA turnover. Biochem Soc Trans 40(4):856–864. https://doi.org/10.1042/bst20120100
Bryant CD, Yazdani N (2016) RNA-binding proteins, neural development and the addictions. Genes Brain Behav 15(1):169–186. https://doi.org/10.1111/gbb.12273
Buchan JR, Parker R (2009) Eukaryotic stress granules : the ins and out of translation what are stress granules ? Mol Cell 36(6):932–932
Buckanovich RJ, Posner JB, Darnell RB (1993) Nova, the paraneoplastic Ri antigen, is homologous to an RNA-binding protein and is specifically expressed in the developing motor system. Neuron 11(4):657–672. https://doi.org/10.1016/0896-6273(93)90077-5
Burd CG, Dreyfusst G (1994) Conserved structures. Science 265(5172):615–621
Calabretta S, Richard S (2015) Emerging Roles of Disordered Sequences in RNA-Binding Proteins. Trends Biochem Sci 40(11):662–672. https://doi.org/10.1016/j.tibs.2015.08.012
Cao X, Jin X, Liu B (2020) The involvement of stress granules in aging and aging-associated diseases. Aging Cell 19(4):1–20. https://doi.org/10.1111/acel.13136
Celona B et al (2017) Suppression of C9orf72 RNA repeat-induced neurotoxicity by the ALS-associated RNA-binding protein Zfp106. Elife 6. https://doi.org/10.7554/eLife.19032
Chang KY, Ramos A (2005) The double-stranded RNA-binding motif, a versatile macromolecular docking platform. FEBS J 272(9):2109–2117. https://doi.org/10.1111/j.1742-4658.2005.04652.x
Chao Y et al (2021) Regulatory roles and mechanisms of alternative RNA splicing in adipogenesis and human metabolic health. Cell Biosci 11(1):1–16. https://doi.org/10.1186/s13578-021-00581-w
Chen Y, Varani G (2005) Protein families and RNA recognition. FEBS J 272(9):2088–2097. https://doi.org/10.1111/j.1742-4658.2005.04650.x
Cioni JM et al (2019) Late endosomes act as mRNA translation platforms and sustain mitochondria in axons. Cell 176(1–2):56-72.e15. https://doi.org/10.1016/j.cell.2018.11.030
Darnell JC, Richter JD (2012) Cytoplasmic RNA-binding proteins and the control of complex brain function. Cold Spring Harb Perspect Biol 4(8):1–17. https://doi.org/10.1101/cshperspect.a012344
Das S, Singer RH, Yoon Y (2019) The travels of mRNAs in neurons: do they know where they are going?. Physiol Behav 57:110–116. https://doi.org/10.1016/j.conb.2019.01.016.The
De Conti L, Baralle M, Buratti E (2017) Neurodegeneration and RNA-binding proteins. Wiley Interdisciplinary Reviews: RNA 8(2):1–12. https://doi.org/10.1002/wrna.1394
Decker CJ, Parker R (2012) P-bodies and stress granules: possible roles in the control of translation and mRNA degradation. Cold Spring Harb Perspect Biol 4(9):1–16
Deshaies JE et al (2018) TDP-43 regulates the alternative splicing of hnRNP A1 to yield an aggregation-prone variant in amyotrophic lateral sclerosis. Brain 141(5):1320–1333. https://doi.org/10.1093/brain/awy062
Dictenberg JB, Swanger SA, Antar LN, Singer RH, Bassell GJ (2008) A direct role for FMRP in activity-dependent dendritic mRNA transport links filopodial-spine morphogenesis to fragile X syndrome. Dev Cell 14(6):926–939. https://doi.org/10.1016/j.devcel.2008.04.003
Duan Y et al (2019) PARylation regulates stress granule dynamics, phase separation, and neurotoxicity of disease-related RNA-binding proteins. Cell Res 29(3):233–247. https://doi.org/10.1038/s41422-019-0141-z
Duszczyk M et al (2019) The solution structure of Dead End bound to AU-rich RNA reveals an unprecedented mode of tandem RRM-RNA recognition required for mRNA regulation. BioRxiv 572156–572156. https://doi.org/10.1101/572156
Ebstein SY, Yagudayeva I, Shneider NA (2019) Mutant TDP-43 causes early-stage dose-dependent motor neuron degeneration in a TARDBP knockin mouse model of ALS. Cell Rep 26(2):364-373.e4. https://doi.org/10.1016/j.celrep.2018.12.045
Eliscovich C, Singer RH (2017) RNP transport in cell biology: the long and winding road. Curr Opin Cell Biol 45:38–46. https://doi.org/10.1016/j.ceb.2017.02.008
Erkelenz S et al (2013) Position-dependent splicing activation and repression by SR and hnRNP proteins rely on common mechanisms. RNA 19(1):96–102. https://doi.org/10.1261/rna.037044.112
Fan XC, Steitz JA (1998) HNS, a nuclear-cytoplasmic shuttling sequence in HuR (nuclear localization͞RNA degradation͞nuclear export). Biochemistry 95:15293–15298
Fay MM, Anderson PJ, Ivanov P (2017) ALS/FTD-associated C9ORF72 repeat RNA promotes phase transitions in vitro and in cells. Cell Rep 21(12):3573–3584. https://doi.org/10.1016/j.celrep.2017.11.093
Feiguin F, Godena VK, Romano G, D’Ambrogio A, Klima R, Baralle FE (2009) Depletion of TDP-43 affects Drosophila motoneurons terminal synapsis and locomotive behavior. FEBS Lett 583(10):1586–1592. https://doi.org/10.1016/j.febslet.2009.04.019
Filippova N et al (2017) Hu antigen R (HuR) multimerization contributes to glioma disease progression. J Biol Chem 292(41):16999–17010. https://doi.org/10.1074/jbc.M117.797878
Fredericks AM, Cygan KJ, Brown BA, Fairbrother WG (2015) RNA-binding proteins: Splicing factors and disease. Biomolecules 5(2):893–909. https://doi.org/10.3390/biom5020893
Gallagher C, Ramos A (2018) Joining the dots – protein-RNA interactions mediating local mRNA translation in neurons. FEBS Lett 592(17):2932–2947. https://doi.org/10.1002/1873-3468.13121
García-Mauriño SM et al (2017) RNA binding protein regulation and cross-talk in the control of AU-rich mRNA Fate. Front Mol Biosci 4:1–9. https://doi.org/10.3389/fmolb.2017.00071
Garcia-Moreno M et al (2019) System-wide profiling of RNA-binding proteins uncovers key regulators of virus infection. Mol Cell 74(1):196-211.e11. https://doi.org/10.1016/j.molcel.2019.01.017
Gerstberger S, Hafner M, Tuschl T (2014) A census of human RNA-binding proteins. Nat Rev Genet 15(12):829–845. https://doi.org/10.1038/nrg3813
Geuens T, Bouhy D, Timmerman V (2016) The hnRNP family: insights into their role in health and disease. Hum Genet 135(8):851–867. https://doi.org/10.1007/s00439-016-1683-5
Glisovic T, Bachorik JL, Yong J, Dreyfuss G (2008) RNA-binding proteins and post-transcriptional gene regulation. FEBS Lett 582(14):1977–1986. https://doi.org/10.1016/j.febslet.2008.03.004
Goldstrohm AC, Hall TMT, McKenney KM (2018) Post-transcriptional regulatory functions of mammalian Pumilio proteins. Trends Genet 34(12):972–990. https://doi.org/10.1016/j.tig.2018.09.006
Gowell M, Baker I, Ansorge O, Husain M (2021) Young-onset frontotemporal dementia with FUS pathology. Pract Neurol 21(2):149–152. https://doi.org/10.1136/practneurol-2020-002730
Grammatikakis I, Abdelmohsen K, Gorospe M (2017) Posttranslational control of HuR function. Wiley Interdisciplinary Reviews: RNA 8:(1). https://doi.org/10.1002/wrna.1372
Gregory JM, Fagegaltier D, Phatnani H, Harms MB (2020) Genetics of amyotrophic lateral sclerosis. Curr Gen Med Rep 8(5):21–131. https://doi.org/10.1101/cshperspect.a024125
Grooms SY et al (2006) Activity bidirectionally regulates AMPA receptor mRNA abundance in dendrites of hippocampal neurons. J Neurosci 26(32):8339–8351. https://doi.org/10.1523/jneurosci.0472-06.2006
Guvenek A, Tian B (2018) Analysis of alternative cleavage and polyadenylation in mature and differentiating neurons using RNA-seq data. Quant Biol 6(3):253–266. https://doi.org/10.1007/s40484-018-0148-3
Habchi J, Tompa P, Longhi S, Uversky VN (2014) Introducing protein intrinsic disorder. Chem Rev 114(13):6561–6588. https://doi.org/10.1021/cr400514h
Hake LE, Mendez R, Richter JD (1998) Specificity of RNA Binding by CPEB: Requirement for RNA Recognition Motifs and a Novel Zinc Finger. Mol Cell Biol 18(2):685–693. https://doi.org/10.1128/mcb.18.2.685
Halbeisen RE, Galgano A, Scherrer T, Gerber AP (2008) Post-transcriptional gene regulation: from genome-wide studies to principles. Cell Mol Life Sci 65(5):798–813. https://doi.org/10.1007/s00018-007-7447-6
Hall DA, Berry-Kravis E (2018) Fragile X syndrome and fragile X-associated tremor ataxia syndrome, 1 ed. (Handbook of Clinical Neurology). Elsevier BV 377–391
Hamada N et al (2016) Essential role of the nuclear isoform of RBFOX1, a candidate gene for autism spectrum disorders, in the brain development. Sci Rep 6(July):1–19. https://doi.org/10.1038/srep30805
Han N, Li W, Zhang M (2013) The function of the RNA-binding protein hnRNP in cancer metastasis. J Cancer Res Ther 9(SUPPL):3. https://doi.org/10.4103/0973-1482.122506
Hanson KA, Kim SH, Wassarman DA, Tibbetts RS (2010) Ubiquilin modifies TDP-43 toxicity in a Drosophila model of amyotrophic lateral sclerosis (ALS). J Biol Chem 285(15):11068–11072. https://doi.org/10.1074/jbc.C109.078527
Hardy RJ (1998) QKI expression is regulated during neuron-glial cell fate decisions. J Neurosci Res 54(1):46–57. https://doi.org/10.1002/(sici)1097-4547(19981001)54:1%3c46::Aid-jnr6%3e3.0.Co;2-h
Hayakawa-Yano Y et al (2017) An RNA-binding protein, Qki5, regulates embryonic neural stem cells through pre-mRNA processing in cell adhesion signaling. Genes Dev 31(18):1910–1925. https://doi.org/10.1101/gad.300822.117.6
Hinnebusch AG, Lorsch JR (2012) The mechanism of eukaryotic translation initiation: New insights and challenges. Cold Spring Harb Perspect Biol 4(10):1–25. https://doi.org/10.1101/cshperspect.a011544
Hofmann S, Kedersha N, Anderson P, Ivanov P (2020) Molecular mechanisms of stress granule assembly and disassembly. Biochimica Et Biophysica Acta - Molecular Cell Research 1868(1):118876–118876. https://doi.org/10.1016/j.bbamcr.2020.118876
Hubstenberger A et al (2017) P-body purification reveals the condensation of repressed mRNA regulons. Mol Cell 68(1):144-157.e5. https://doi.org/10.1016/j.molcel.2017.09.003
Humphrey J et al (2020) FUS ALS-causative mutations impair FUS autoregulation and splicing factor networks through intron retention. Nucleic Acids Res 48(12):6889–6905. https://doi.org/10.1093/nar/gkaa410
Huttelmaier S et al (2005) Spatial regulation of beta-actin translation by Src-dependent phosphorylation of ZBP1. Nature 438(7067):512–515. https://doi.org/10.1038/nature04115
Ince-dunn G et al (2012) Article neuronal Elav-like ( Hu ) proteins regulate RNA splicing and abundance to control glutamate levels and neuronal excitability. Neuron 75(6):1067–1080. https://doi.org/10.1016/j.neuron.2012.07.009
Ivshina M, Lasko P, Richter JD (2014) Cytoplasmic polyadenylation element binding proteins in development, health, and disease. Annu Rev Cell Dev Biol 30:393–415. https://doi.org/10.1146/annurev-cellbio-101011-155831
Jain S, Wheeler JR, Walters RW, Agrawal A, Barsic A, Parker R (2016) ATPase-modulated stress granules contain a diverse proteome and substructure. Cell 164(3):487–498. https://doi.org/10.1016/j.cell.2015.12.038
Jankowsky E, Harris ME (2015) Specificity and nonspecificity in RNA-protein interactions. Nat Rev Mol Cell Biol 16(9):533–544. https://doi.org/10.1038/nrm4032
Janssens J et al (2013) Overexpression of ALS-associated p. M337V human TDP-43 in mice worsens disease features compared to wild-type human TDP-43 mice. Mol Neurobiol 48(1):22–35. https://doi.org/10.1007/s12035-013-8427-5
Jin Y et al (2003) A vertebrate RNA-binding protein Fox-1 regulates tissue-specific splicing via the pentanucleotide GCAUG. EMBO J 22(4):905–912. https://doi.org/10.1093/emboj/cdg089
Jo M, Lee S, Jeon YM, Kim S, Kwon Y, Kim HJ (2020) The role of TDP-43 propagation in neurodegenerative diseases: integrating insights from clinical and experimental studies. Exp Mol Med 52(10):1652–1662. https://doi.org/10.1038/s12276-020-00513-7
Jung H, Gkogkas CG, Sonenberg N, Holt CE (2014) Remote control of gene function by local translation. Cell 157(1):26–40. https://doi.org/10.1016/j.cell.2014.03.005
Kanopka A, Muhlemann O, Akusjarvi G (1996) Inhibition by SR proteins splicing of a regulated adenovirus pre-mRNA. Nature 381:535–538
Kapeli K, Martinez FJ, Yeo GW (2017) Genetic mutations in RNA-binding proteins and their roles in ALS. Hum Genet 136(9):1193–1214. https://doi.org/10.1007/s00439-017-1830-7
Kedersha N et al (2000) Dynamic shuttling of TIA-1 accompanies the recruitment of mRNA to mammalian stress granules. J Cell Biol 151(6):1257–1268. https://doi.org/10.1083/jcb.151.6.1257
Keene JD (2001) Ribonucleoprotein infrastructure regulating the flow of genetic information between the genome and the proteome. Proc Natl Acad Sci USA 98(13):7018–7024. https://doi.org/10.1073/pnas.111145598
Kim SH, Zhan L, Hanson KA, Tibbetts RS (2012) High-content RNAi screening identifies the type 1 inositol triphosphate receptor as a modifier of TDP-43 localization and neurotoxicity. Hum Mol Genet 21(22):4845–4856. https://doi.org/10.1093/hmg/dds321
Kohrmann M, Luo M, Kaether C, DesGroseillers L, Dotti CG, Kiebler MA (1999) Microtubule-dependent recruitment of Staufen-green fluorescent protein into large RNA-containing granules and subsequent dendritic transport in living hippocampal neurons. Mol Biol Cell 10(9):2945–2953. https://doi.org/10.1091/mbc.10.9.2945
Kozlov E, Shidlovskii YV, Gilmutdinov R, Schedl P, Zhukova M (2021) The role of CPEB family proteins in the nervous system function in the norm and pathology. Cell Biosci 11(1):1–14. https://doi.org/10.1186/s13578-021-00577-6
Krichevsky AM, Kosik KS (2001) Neuronal RNA granules: a link between RNA localization and stimulation-dependent translation. Neuron 32(4):683–696. https://doi.org/10.1016/s0896-6273(01)00508-6
Kuo PH, Chiang CH, Wang YT, Doudeva LG, Yuan HS (2014) The crystal structure of TDP-43 RRM1-DNA complex reveals the specific recognition for UG- and TG-rich nucleic acids. Nucleic Acids Res 42(7):4712–4722. https://doi.org/10.1093/nar/gkt1407
Lauriat TL et al (2008) Developmental expression profile of Quaking, a candidate gene for schizophrenia, and its target genes in human prefrontal cortex and hippocampus shows regional specificity. J Neurosci Res 86(4):785–796. https://doi.org/10.1002/jnr.21534
Lee J-A et al (2016) Cytoplasmic Rbfox1 regulates the expression of synaptic and autism-related genes. Neuron 89(1):113–128. https://doi.org/10.1016/j.neuron.2015.11.025.Cytoplasmic
Lee Y, Rio DC, Biology S, Biology C (2015) Mechanisms and Regulation of Alternative Pre-mRNA Splicing Yeon. Annu Rev Biochem (258):291–323. https://doi.org/10.1146/annurev-biochem-060614-034316.Mechanisms
Lewis HA et al (2000) Sequence-specific RNA binding by a Nova KH domain: implications for paraneoplastic disease and the fragile X syndrome. Cell 100(3):323–332. https://doi.org/10.1016/s0092-8674(00)80668-6
Licatalosi DD et al (2008) HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature 456(7221):464–469. https://doi.org/10.1038/nature07488
Lukong KE, Chang KW, Khandjian EW, Richard S 2008) RNA-binding proteins in human genetic disease. Trends Gen 24(8):416–425. https://doi.org/10.1016/j.tig.2008.05.004
Lunde BM, Moore C, Varani G (2007) RNA-binding proteins: Modular design for efficient function. Nat Rev Mol Cell Biol 8(6):479–490. https://doi.org/10.1038/nrm2178
Malik AM, Miguez RA, Li X, Ho Y-S, L EF, Barmada SJ (2018) Matrin 3-dependent neurotoxicity is modified by nucleic acid binding and nucleocytoplasmic localization. BioRxiv 1–52
Mann JR et al (2019) RNA binding antagonizes neurotoxic phase transitions of TDP-43. Neuron 102(2):321–338 e8. https://doi.org/10.1016/j.neuron.2019.01.048
Maris C, Dominguez C, Allain FHT (2005) The RNA recognition motif, a plastic RNA-binding platform to regulate post-transcriptional gene expression. FEBS J 272(9):2118–2131. https://doi.org/10.1111/j.1742-4658.2005.04653.x
Markmiller S et al (2018) Context-dependent and disease-specific diversity in protein interactions within stress granules. Cell 172(3):590-604.e13. https://doi.org/10.1016/j.cell.2017.12.032
Martin KC, Ephrussi A (2009) mRNA localization: gene expression in the spatial dimension. Cell 136(4):719–730. https://doi.org/10.1016/j.cell.2009.01.044
Martin S, Tazi J (2014) Visualization of G3BP stress granules dynamics in live primary cells. J vis Exp 43(87):1–8. https://doi.org/10.3791/51197
Martinez-Contreras R, Fisette JF, Nasim FUH, Madden R, Cordeau M, Chabot B (2006) Intronic binding sites for hnRNP A/B and hnRNP F/H proteins stimulate pre-mRNA splicing. PLoS Biol 4(2):172–185. https://doi.org/10.1371/journal.pbio.0040021
Masuda A et al (2015) Position-specific binding of FUS to nascent RNA regulates mRNA length. Genes Dev 29(10):1045–1057. https://doi.org/10.1101/gad.255737.114
Matoulkova E, Michalova E, Vojtesek B, Hrstka AR (2012) The role of the 3' untranslated region in post-transcriptional regulation of protein expression in mammalian cells. RNA Biol 9(5):563–576
Moore KS, von Lindern M (2018) RNA binding proteins and regulation of mRNA translation in erythropoiesis. Front Physiol 9:1–17. https://doi.org/10.3389/fphys.2018.00910
Moore S et al (2019) ADAR2 mislocalization and widespread RNA editing aberrations in C9orf72-mediated ALS/FTD. Acta Neuropathol 138:49–65. https://doi.org/10.1007/s00401-019-01999-w
Neelagandan N, Lamberti I, Carvalho HJF, Gobet C, Naef F (2020) What determines eukaryotic translation elongation: Recent molecular and quantitative analyses of protein synthesis: Determinants of eukaryotic translation. Open Biol 10(12). https://doi.org/10.1098/rsob.200292rsob200292
Neumann M et al (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314(5796):130–133. https://doi.org/10.1126/science.1134108
Nonhoff et al (2007) Ataxin-2 interacts with the DEAD/H-Box RNA Helicase DDX6 and interferes with P-bodies and stress granules. Mol Biol Cell 18:1385–1396. https://doi.org/10.1091/mbc.E06
Norbury CJ (2013) Cytoplasmic RNA: A case of the tail wagging the dog. Nat Rev Mol Cell Biol 14(10):643–653. https://doi.org/10.1038/nrm3645
Oliveira C, Faoro H, Alves LR, Goldenberg S (2017) RNA-binding proteins and their role in the regulation of gene expression in trypanosoma cruzi and saccharomyces cerevisiae. Genet Mol Biol 40(1):22–30. https://doi.org/10.1590/1678-4685-gmb-2016-0258
Ostrowski LA, Hall AC, Mekhail K (2017) Ataxin-2: from RNA control to human health and disease. Genes 8(6):2–21. https://doi.org/10.3390/genes8060157
Oubridge C, Ito N, Evans PR, Teo CH, Nagai K (1994) Crystal structure at 1.92 Å resolution of the RNA-binding domain of the U1A spliceosomal protein complexed with an RNA hairpin. Nature 372:432–438
Pan X, Fang Y, Li X, Yang Y, Shen HB (2020) RBPsuite: RNA-protein binding sites prediction suite based on deep learning. BMC Genomics 21(1):1–8. https://doi.org/10.1186/s12864-020-07291-6
Panas MD, Ivanov P, Anderson P (2016) Mechanistic insights into mammalian stress granule dynamics. J Cell Biol 215(3):313–323. https://doi.org/10.1083/jcb.201609081
Park E et al (2011) Regulatory roles of heterogeneous nuclear ribonucleoprotein M and Nova-1 protein in alternative splicing of dopamine D2 receptor pre-mRNA. J Biol Chem 286(28):25301–25308. https://doi.org/10.1074/jbc.M110.206540
Parker R, Sheth U (2007) P bodies and the control of mRNA translation and degradation. Mol Cell 25(5):635–646. https://doi.org/10.1016/j.molcel.2007.02.011
Parras A et al (2018) Autism-like phenotype and risk gene mRNA deadenylation by CPEB4 mis-splicing. Genes Dev 560(7719):441–446. https://doi.org/10.1038/s41586-018-0423-5
Pesiridis GS, Lee VMY, Trojanowski JQ (2009) Mutations in TDP-43 link glycine-rich domain functions to amyotrophic lateral sclerosis. Hum Mol Genet 18(R2):156–162. https://doi.org/10.1093/hmg/ddp303
Pilaz LJ, Silver DL (2015) Post-transcriptional regulation in corticogenesis: How RNA-binding proteins help build the brain. Wiley Interdisciplinary Reviews: RNA 6(5):501–515. https://doi.org/10.1002/wrna.1289
Poulopoulos A et al (2019) Subcellular transcriptomes and proteomes of developing axon projections in the cerebral cortex. Nature 565(7739):356–360. https://doi.org/10.1038/s41586-018-0847-y
Raj B, Blencowe BJ (2015) Alternative Splicing in the Mammalian Nervous System: Recent Insights into Mechanisms and Functional Roles. Neuron 87(1):14–27. https://doi.org/10.1016/j.neuron.2015.05.004
Rajman M et al (2017) A microRNA-129-5p/Rbfox crosstalk coordinates homeostatic downscaling of excitatory synapses. EMBO J 36(12):1770–1787. https://doi.org/10.15252/embj.201695748
Rayman JB, Kandel ER (2017) TIA-1 is a functional prion-like protein. 1–13
Richter JD (2007) CPEB: a life in translation. Trends Biochem Sci 32(6):279–285. https://doi.org/10.1016/j.tibs.2007.04.004
Rissland OS et al (2017) The influence of microRNAs and poly(A) tail length on endogenous mRNA-protein complexes. Genome Biol 18(1):1–18. https://doi.org/10.1186/s13059-017-1330-z
Robberecht W, Eykens C (2015) The genetic basis of amyotrophic lateral sclerosis: recent breakthroughs. Adv Genom Gen 327–327. https://doi.org/10.2147/agg.s57397
Schieweck R, Ninkovic J, Kiebler MA (2021) RNA-binding proteins balance brain function in health and disease. Physiol Rev 101(3):1309–1370. https://doi.org/10.1152/physrev.00047.2019
Schoser B, Timchenko L (2010) Myotonic dystrophies 1 and 2: complex diseases with complex mechanisms. Curr Genomics 11(2):77–90. https://doi.org/10.2174/138920210790886844
Schwartz JL, Jones KL, Yeo GW (2021) Repeat RNA expansion disorders of the nervous system: post-transcriptional mechanisms and therapeutic strategies. Crit Rev Biochem Mol Biol 56(1):31–53. https://doi.org/10.1080/10409238.2020.1841726
Selvanathan SP, Graham GT, Erkizan HV, Dirsken U, Natarajan T, and Dakic A (2015) Oncogenic fusion protein EWS-FLI1 is a network hub that regulates alternative splicing. PNAS 12(11)
Sena RM, Twiss JL, Gardiner AS, Dell’orco M, Linsenbardt DN, Perrone-Bizzozero NI (2021) The RNA-binding protein HuD regulates alternative splicing and alternative polyadenylation in the mouse neocortex. Molecules 26(10). https://doi.org/10.3390/molecules26102836
Shashidharan P et al (1997) Immunohistochemical localization of the neuron-specific glutamate transporter EAAC1 (EAAT3) in rat brain and spinal cord revealed by a novel monoclonal antibody. Brain Res 773(1–2):139–148. https://doi.org/10.1016/s0006-8993(97)00921-9
Shaw DJ, Morse R, Todd AG, Eggleton P, Lorson CL, Young PJ (2010) Identification of a self-association domain in the Ewing’s sarcoma protein: a novel function for arginine-glycine-glycine rich motifs? J Biochem 147(6):885–893. https://doi.org/10.1093/jb/mvq025
Shotwell CR, Cleary JD, Berglund JA (2020) The potential of engineered eukaryotic RNA-binding proteins as molecular tools and therapeutics. Wiley Interdisciplinary Reviews: RNA 11(1):1–21. https://doi.org/10.1002/wrna.1573
Shukla S, Parker R (2016) Hypo- and hyper-assembly diseases of RNA–protein complexes. Trends Mol Med 22(7):615–628. https://doi.org/10.1016/j.molmed.2016.05.005
Sibley CR, Blazquez L, Ule J (2016) Lessons from non-canonical splicing Christopher. 17(7)407–421. https://doi.org/10.1038/nrg.2016.46.Lessons
Singh G, Pratt G, Yeo GW, Moore MJ (2015) The clothes make the mRNA: past and present trends in mRNP fashion. Annu Rev Biochem 84(February):325–354. https://doi.org/10.1146/annurev-biochem-080111-092106
Singh RK, Cooper TA (2012) Pre-mRNA splicing in disease and therapeutics. Trends Mol Med 18(8):472–482. https://doi.org/10.1016/j.molmed.2012.06.006
Soheilypour M, Mofrad MRK (2018) Quality control of mRNAs at the entry of the nuclear pore: cooperation in a complex molecular system. Nucleus 9(1):202–211. https://doi.org/10.1080/19491034.2018.1439304
Sonenberg N, Hinnebusch AG (2009) Regulation of translation initiation in eukaryotes. Cell 136(4):731–745. https://doi.org/10.1016/j.cell.2009.01.042.Regulation
Song T et al (2015) Specific interaction of KIF11 with ZBP1 regulates the transport of beta-actin mRNA and cell motility. J Cell Sci 128(5):1001–1010. https://doi.org/10.1242/jcs.161679
Sreedharan J et al (2008) TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319(5870):1668–1672. https://doi.org/10.1126/science.1154584
Stefl R, Skrisovska L, Allain FHT (2005) RNA sequence- and shape-dependent recognition by proteins in the ribonucleoprotein particle. EMBO Rep 6(1):33–38. https://doi.org/10.1038/sj.embor.7400325
Suresh S, Joladarashi D, Jeyabal P, Amirthalingam R, Krishnamurthy TA (2016) RNA stabilizing proteins as molecular targets in cardiovascular pathologies. Trends Cardiovasc Med 176(1):139–148
Szostak E, Gebauer F (2013) Translational control by 3’-UTR-binding proteins. Brief Funct Genomics 12(1):58–65. https://doi.org/10.1093/bfgp/els056
Telias M (2019) Molecular mechanisms of synaptic dysregulation in fragile X syndrome and autism spectrum disorders. Front Mol Neurosci 12(March):1–12. https://doi.org/10.3389/fnmol.2019.00051
Terenzio M et al (2018) Locally translated mTOR controls axonal local translation in nerve injury. 1421:1416–1421
Thelen K, Maier B, Faber M, Albrecht C, Fischer P, Pollerberg GE (2012) Translation of the cell adhesion molecule ALCAM in axonal growth cones - regulation and functional importance. J Cell Sci 125(Pt 4):1003–1014. https://doi.org/10.1242/jcs.096149
Thelen MP, Kye MJ (2020) The role of RNA binding proteins for local mRNA translation: implications in neurological disorders. Front Mol Biosci 6(January):1–13. https://doi.org/10.3389/fmolb.2019.00161
Tiruchinapalli DM et al (2003) Activity-dependent trafficking and dynamic localization of zipcode binding protein 1 and β-actin mRNA in dendrites and spines of hippocampal neurons. J Neurosci 23(8):3251–3261. https://doi.org/10.1523/jneurosci.23-08-03251.2003
Toll-Riera M, Radó-Trilla N, Martys F, Albá MM (2012) Role of low-complexity sequences in the formation of novel protein coding sequences. Mol Biol Evol 29(3):883–886. https://doi.org/10.1093/molbev/msr263
Turner-Bridger B, Caterino C, Cioni JM (2020) Molecular mechanisms behind mRNA localization in axons: axonal mRNA localisation. Open Biol 10(9). https://doi.org/10.1098/rsob.200177
Udan M, Baloh RH (2011) Implications of the prion-related Q/N domains in TDP-43 and FUS. Prion 5(1):1–5. https://doi.org/10.4161/pri.5.1.14265
Ule J et al (2006) An RNA map predicting Nova-dependent splicing regulation. Nature 444(7119):580–586. https://doi.org/10.1038/nature05304
Uversky VN (2019) Intrinsically disordered proteins and their “Mysterious” (meta)physics. Front Phys 7:8–23. https://doi.org/10.3389/fphy.2019.00010
Valverde R, Edwards L, Regan L (2008) Structure and function of KH domains. FEBS J 275(11):2712–2726. https://doi.org/10.1111/j.1742-4658.2008.06411.x
Van Nostrand EL et al (2020) A large-scale binding and functional map of human RNA-binding proteins. Nature 583(7818):711–719. https://doi.org/10.1038/s41586-020-2077-3
Vance C et al (2009) Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323(5918):1208–1211. https://doi.org/10.1126/science.1165942
Vanderweyde T, Youmans K, Liu-Yesucevitz L, Wolozin B (2013) Role of stress granules and RNA-binding proteins in neurodegeneration: a mini-review. Gerontology 59(6):524–533. https://doi.org/10.1159/000354170
Varani G, Nagai K (1998) RNA recognition by RNP proteins during RNA processing. Annu Rev Biophys Biomol Struct 27:407–445. https://doi.org/10.1146/annurev.biophys.27.1.407
Wang I, Hennig J, Jagtap PKA, Sonntag M, Valcárcel J, Sattler M (2014) Structure, dynamics and RNA binding of the multi-domain splicing factor TIA-1. Nucleic Acids Res 42(9):5949–5966. https://doi.org/10.1093/nar/gku193
Wang et al (2015) Mechanism of alternative splicing and its regulation. Biomedical Reports 3(2):152–158. https://doi.org/10.3892/br.2014.407
Wang ET et al (2016) Dysregulation of mRNA localization and translation in genetic disease. J Neurosci 36(45):11418–11426. https://doi.org/10.1523/JNEUROSCI.2352-16.2016
Wang Z et al (2019) The emerging roles of hnRNPK. J Cell Physiol 235(3):1995–2008. https://doi.org/10.1002/jcp.29186
Watanabe R et al (2020) Intracellular dynamics of Ataxin-2 in the human brains with normal and frontotemporal lobar degeneration with TDP-43 inclusions. Acta Neuropathol Commun 8(1):1–19. https://doi.org/10.1186/s40478-020-01055-9
Wegorzewska I, Bell S, Cairns NJ, Miller TM, Baloh RH (2009) TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci USA 106(44):18809–18814. https://doi.org/10.1073/pnas.0908767106
Wolfe SA, Nekludova L, Pabo CO (2000) DNA RECOGNITION BY Cys2His2 ZINC FINGER PROTEINS. Annual Review of Biophysics and Biomolecular Structure Rev Biophys Biomol Struct 3:183–212
Wolozin and Ivanov (2019) Stress granules and neurodegeneration. Nat Rev Neurosci 20(11):649–666. https://doi.org/10.1038/s41583-019-0222-5.Stress
Workman E, Veith A, Battle DJ (2014) U1A Regulates 3’ processing of the survival motor neuron mRNA. J Biol Chem 289(6):3703–3712. https://doi.org/10.1074/jbc.M113.538264
Xie W, Denman RB (2011) Protein methylation and stress granules: posttranslational remodeler or innocent bystander? Molecular Biology International 2011:1–14. https://doi.org/10.4061/2011/137459
Xing Y et al (2021) Downregulation of NUDT21 contributes to cervical cancer progression through alternative polyadenylation. Oncogene 40(11):2051–2064. https://doi.org/10.1038/s41388-021-01693-w
Xu Y et al (2019) Post-translational modification control of RNA-binding protein hnRNPK function. Open Biol 9(3). https://doi.org/10.1098/rsob.180239
Xu YF et al (2010) Wild-type human TDP-43 expression causes TDP-43 phosphorylation, mitochondrial aggregation, motor deficits, and early mortality in transgenic mice. J Neurosci 30(32):10851–10859. https://doi.org/10.1523/jneurosci.1630-10.2010
Yang YM et al (2018) Identification of a molecular locus for normalizing dysregulated GABA release from interneurons in the Fragile X brain. Mol Psychiatry 25(9):2017–2035. https://doi.org/10.1038/s41380-018-0240-0
Yang YYL, Yin GL, Darnell RB (1998) The neuronal RNA-binding protein Nova-2 is implicated as the autoantigen targeted in POMA patients with dementia. Proc Natl Acad Sci USA 95(22):13254–13259. https://doi.org/10.1073/pnas.95.22.13254
Yee BA, Pratt GA, Graveley BR, van Nostrand EL, Yeo GW (2019) RBP-Maps enables robust generation of splicing regulatory maps. RNA 25(2):193–204. https://doi.org/10.1261/rna.069237.118
Yu Y, Li Y, Zhang Y (2015) Screening of APP interaction proteins by DUALmembrane yeast two-hybrid system. Int J Clin Exp Pathol 8(3):2802–2808
Zheng X et al (2020) Serine/arginine-rich splicing factors: The bridge linking alternative splicing and cancer. Int J Biol Sci 16(13):2442–2453. https://doi.org/10.7150/ijbs.46751
Zhong YI, Hu Z, Wu J, Dai FAN, Lee F, Xu Y (2020) STAU1 selectively regulates the expression of inflammatory and immune response genes and alternative splicing of the nerve growth factor receptor signaling pathway. Oncol Rep 44(5):1863–1874. https://doi.org/10.3892/or.2020.7769
Zhou B et al (2020) FUS P525L mutation causing amyotrophic lateral sclerosis and movement disorders. Brain and Behavior 10(6):1–9. https://doi.org/10.1002/brb3.1625
Zhou HL, Mangelsdorf M, Liu JH, Zhu L, Wu JY (2014) RNA-binding proteins in neurological diseases. Science China Life Sciences 57(4):432–444. https://doi.org/10.1007/s11427-014-4647-9
Zhu H, Zhou HL, Hasman RA, Lou H (2007) Hu proteins regulate polyadenylation by blocking sites containing U-rich sequences. J Biol Chem 282(4):2203–2210. https://doi.org/10.1074/jbc.M609349200
Acknowledgements
All figures were created with Adobe Illustrator.
Funding
This review was supported by grants from the Consejo Nacional de Ciencia y Tecnología (Conacyt, México): Ph.D. scholarship 779191 (AO-M), Postdoctoral research grant 2753117 (JL-L), 255087 (AO).
Author information
Authors and Affiliations
Contributions
Arturo Ortega: conceptualization and writing of the final manuscript. Andrea Ocharán-Mercado: conceptualization and writing original draft preparation. Jaqueline Loaeza-Loaeza: writing—original draft preparation and figures preparation. Daniel Hernández-Sotelo, Yaneth Castro-Coronel, Leonor C Acosta Saavedra, Luisa C Hernández-Kelly: reviewing and editing. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ocharán-Mercado, A., Loaeza-Loaeza, J., Castro-Coronel, Y. et al. RNA-Binding Proteins: A Role in Neurotoxicity?. Neurotox Res 41, 681–697 (2023). https://doi.org/10.1007/s12640-023-00669-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12640-023-00669-w