
Abstract
Cardiovascular disease (CVD) has reached epidemic proportions and is a leading cause of death worldwide. One of the long-standing goals of scientists is to repair heart tissue damaged by various forms of CVD such as cardiac hypertrophy, dilated cardiomyopathy, myocardial infarction, heart fibrosis, and genetic and developmental heart defects such as heart valve deformities. Damaged or defective heart tissue has limited regenerative capacity and results in a loss of functioning myocardium. Advances in transcriptomic profiling technology have revealed that long noncoding RNA (lncRNA) is transcribed from what was once considered “junk DNA.” It has since been discovered that lncRNAs play a critical role in the pathogenesis of various CVDs and in myocardial regeneration. This review will explore how lncRNAs impact various forms of CVD as well as those involved in cardiomyocyte regeneration. Further, we discuss the potential of lncRNAs as a therapeutic modality for treating CVD.
Similar content being viewed by others

Introduction
The Human Genome Project started in 2001 [1] and was 99% completed in 2004 [2], and during this time it became clear that only approximately 1.5% of the human genome coded for approximately 21,000 genes [3]. Non-coding DNA, which does not code for a protein, was termed “junk DNA” [4]; however, with the development of genome tiling array technology, The Encyclopedia of DNA Elements (ENCODE) project in 2012 found that RNA transcripts arose from at least 76% of the human genome and these RNA molecules likely had biological functions [5]. According to the Human GENCODE project, there are more than 16,000 currently identified long noncoding RNAs (lncRNAs) [6] defined as RNAs that do not code for proteins and are longer than 200 nucleotides in length [7]. lncRNAs are primarily transcribed by RNA polymerase II and are classified into five types according to their structural location: sense, antisense, bidirectional, intronic, and intergenic lncRNAs [5].
lncRNAs perform multiple functions through various mechanisms. At the transcriptional level, lncRNAs function as signals, decoys, guides, and scaffolds. By influencing signaling factors or acting by themselves, lncRNAs can regulate downstream genes. lncRNAs can also negatively regulate downstream genes by decoying protein molecules to block specific molecular pathways, acting as guides for the localization of specific molecules to particular locations, and/or functioning as scaffolding that favors the association of various proteins to promote function as a macromolecular complex [5, 8]. At the post-transcriptional level, lncRNAs can regulate mRNA splicing by binding to proteins that modulate mRNA turnover, and/or stabilize and translate mRNA transcripts by binding to target RNA. lncRNAs also play a role in sponging miRNA, with sequestration of microRNAs, and serve as competing endogenous RNA (ceRNA) to regulate mRNA expression [7, 9]. Transcriptional genome-wide studies (TWAS) have found that lncRNAs contribute to both human disease and genetic traits. TWAS and co-localization analysis identified 14,100 lncRNAs from 49 tissues contributing to 101 genetic traits [10]. It has been reported that lncRNAs regulate gene action during cardiovascular disease (CVD) and cardiac development. Examining how lncRNAs are produced and regulated in disease states is crucial to finding clues for novel approaches to disease treatment.
CVD is a condition in which either heart or blood vessels are structurally or functionally abnormal. Although numerous pharmacological and interventional therapies for CVD have been developed to date, and the World Health Organization (WHO) has instituted the “Global Action Plan for the Prevention and Control of Non-communicable Diseases (NCDs) 2013–2020” mission, CVD remains a leading cause of morbidity and mortality worldwide [11]. According to National Health and Nutrition Examination Survey (NHANES) data, the prevalence of CVD in adults aged 20 years or older was 49.2% [12]. WHO estimates that 17.9 million people died from CVD, which represents 32% of all deaths worldwide, and 37% of non-communicable diseases among those under 70 years of age in 2019 [13]. The Global Burden Disease (GBD) 2020 study shows an estimated 19.05 million people died in 2020 from CVD, an 18.71% increase from 2010 [14].
By analyzing lncRNAs and understanding their molecular basis for various forms of CVDs such as MI, cardiac fibrosis, and cardiomyopathy (Table 1), as well as analyzing their role in cardiomyocyte regeneration through controlling proliferation and development (Table 2), and by exploring how lncRNAs can be effectively harnessed for the treatment of CVD, we may be able to identify new therapeutic targets for treating this common form of disease.
lncRNAs in failing heart and cardiac regeneration
lncRNAs in myocardial infarction
Myocardial infarction (MI) occurs when the myocardium is deprived of oxygen for a prolonged period of time, resulting in hypoperfusion and necrosis, which can result in sudden death. Due to its sudden onset, MI is also referred to as “acute” MI [15]. According to surveys conducted by the Atherosclerosis Risk in Communities (ARIC) study between 2005 and 2014, there are 605,000 new cases of MI each year and 200,000 cases of recurrent MI. Furthermore, the MI incidence rate is higher in men than in women with an age of first onset of 65.5 years in men and 72.0 years in women [16]. Certain data indicate a higher occurrence of MI in men, attributed to differences in lifestyle and customs. Men often have more demanding occupations and employ distinct stress management techniques compared to women. They may also pay less attention to maintaining a healthy diet, have a higher likelihood of being overweight, and might be less mindful of their own symptoms and well-being [17]. Consequently, when compared to women with similar risk factors, men with hypertension, high BMI (body mass index), and type II diabetes experience higher rates of MI [18]. Although MI is more common in men than in women throughout their lives, the gender differences in MI risk tend to reduce as individuals age [19]. Figure 1 illustrates the mechanisms through which lncRNAs associated with myocardial infarction function.

The regulatory functions involved in myocardial infarction
In the nucleus of cardiomyocytes affected by MI, the expression of lncRNA small nucleolar RNA host gene 1 (Snhg1) increases within in the damaged heart tissue. Snhg1 acts as a signal molecule by decoying phosphatase and tensin homolog (PTEN) to regulate signal pathways. After MI, Snhg1 is mainly expressed in cardiomyocytes and binds to the PTEN protein, resulting in its degradation and consequential activation of PI3K/Akt signaling. c-Myc, a downstream protein in the PI3K/Akt signaling pathway, forms a positive feedback loop with Snhg1 by binding to the Snhg1 promoter. Consequently, Snhg1 regulates cardiomyocyte proliferation after MI by upregulating PI3K/Akt signaling and promoting an angiogenic response through the induction of VEGFA gene expression [20].
Within the cytoplasm, lncRNA can sponge miRNA molecules and thus play an important role in angiogenesis, mitochondrial dynamics, apoptosis, and cell viability by sequestering molecules. The lncRNA AK017121 (UCSC Genome Browser on Mouse, 2011 Assembly) is also termed the cardiac apoptosis-related lncRNA (Carl) and is highly expressed in cardiomyocytes injured following MI ischemia. Carl sponges miR-539 by directly binding to it and this reduces infarct size during MI as miR-539 suppresses prohinin2 (Phb2) expression by binding to Phb2 mRNA and the Phb2 protein functions in inhibiting mitochondrial fission and apoptosis. To put it concisely, Carl regulates mitochondrial dynamics through the Carl/miR-539/Phb2 signaling axis [21, 22].
Another example of an important lncRNA in the heart is cardiac hypertrophy-related factor (Chrf). This lncRNA is expressed not only in cardiac hypertrophy but also in myocardial I/R injury models. In the context of myocardial I/R injury, silencing of Chrf reduces autophagy, inhibits apoptosis, increases cell viability, and reduces lactate dehydrogenase (LDH) levels. Chrf appears to limit myocardial I/R injury by preventing Atg7 from being inhibited by miR-182-5p [23].
The lncRNA Opa-interacting protein 5-antisense transcript 1 (Oip5-as1) is also downregulated in cardiomyocytes in response to oxygen–glucose deprivation/reoxygenation (OGD/R), an in vitro MI injury model. Oip5-as1 plays a role in reducing oxidative stress in OGD/R conditions by acting as a ceRNA and sponging miR-29a and subsequently preventing miR-29a from binding to the 3′ UTR of the sirtuin-1 (Sirt1) mRNA. This action results in the activation of the Sirt1/5′ AMP-activated protein kinase (Ampk)/peroxisome proliferator-activated receptor gamma coactivator 1-alpha (Pgc1-α) pathway. In this pathway, Sirt1 is known to suppress oxidative stress and apoptosis during MI; Ampk is involved in energy metabolism which aids cell survival following ischemic injury; and Pgc1-α is responsible for energy homeostasis, oxidative metabolism, and cardiac mitochondrial function. Such functions of the Sirt1/Ampk/Pgc1-α pathway benefit survival of cardiomyocyte cells during MI injury [24].
HOX antisense intergenic RNA (Hotair) is decreased during acute myocardial infarction (AMI) both in a mouse model and human patient serum. Hotair prevents hypoxia-induced cardiomyocyte apoptosis [25], and since Hotair has a binding site for miR-206, it prevents inactivation of fibronectin 1 (Fn1) by sponging miR-206. The role of Fn1 in cardiac vascular disease has not yet been clearly elucidated, but is known to have low expression in patients with congestive heart failure and is secreted from migrating cardiac valve interstitial cells. Additionally, it is known that the miR-206/Fn1 axis inhibits apoptosis in some forms of cancer. Thus, the Hotair/miR-206/Fn1 axis is similarly thought to prevent apoptosis following AMI [26].
lncRNAs in heart fibrosis
Cardiac fibrosis refers to pathophysiologic event showing increased myofibroblast activity and excessive extracellular matrix accumulation, particularly collagen type I, during cardiac remodeling in most cardiac disease. The principal cause of myocardial interstitial fibrosis is MI but is also observed in hypersensitive heart disease, hypertrophic cardiomyopathy (HCM), and idiopathic dilated cardiomyopathy (DCM) [27]. Cardiac fibrosis can be classified into two main categories: heart failure with reduced ejection fraction (HFrEF) or heart failure with preserved ejection fraction (HFpEF) [28]. In HFrEF, the heart’s ability to generate systolic force is compromised, leading to a decrease in the EF—the proportion of blood expelled with each contraction. On the other hand, in HFpEF, the typical parameters of systolic function are mostly preserved, but diastolic filling and relaxation are impaired [28]. Figure 2 demonstrates the functions of lncRNAs related to heart fibrosis.

The regulatory functions involved in heart fibrosis
In cardiac fibroblasts, some lncRNAs regulate fibrosis-related genes by modulating transcription factor occupancy within gene promoter regions. The lncRNA maternally expressed gene 3 (Meg3) is most abundant in the nucleus of cardiac fibroblasts and its expression is downregulated in mice after transverse aortic constriction (TAC). Meg3 mechanistically decoys p53 by interfering with p53’s ability to bind to the promoter of matrix metallopeptidase 2 (Mmp2) and thus allowing the cell to upregulate Mmp2 expression [29]. As the major role of Mmp2 is extracellular matrix (ECM) remodeling, reduced Meg3 levels help drive Mmp2 expression therefore reducing fibrosis and myocardial hypertrophy as well as improving diastolic function in a mouse model of TAC. Therefore, Meg3 is an attractive target for limiting ECM remodeling during the process of heart fibrosis [30].
Wisp2 super-enhancer-associated RNA (Wisper) is a lncRNA that induces fibrosis by acting as guide molecule in the nucleus of fibroblast. Wisper is highly expressed in MI animal models as well as in human aortic stenosis patients. When Wisper expression is knocked down, cell migration and proliferation decreased, and apoptosis increased when compared to controls. RNA sequencing also revealed that pro-apoptotic genes and cell cycle inhibitors were increased, while pro-inhibitory molecules that promote fibrosis were decreased. Conversely, when Wisper was overexpressed, expression of fibrosis-related genes also increased. Specifically, when Wisper was delivered intraperitoneally into a mouse model of induced MI, cardiac fibrosis was reduced. The mechanism of how Wisper causes fibrosis is that the T-cell intracellular antigen 1 (TIA1)-related/like protein (Tiar) protein binds to Wisper and promotes the expression of heart-specific procollagen-lysine,2-oxoglutarate 5-dioxygenase 2 (Plod2) gene. Plod2 subsequently induces collagen synthesis and makes the extracellular matrix stable. In sum, the identification of Wisper represents crucial step toward anti-fibrotic therapeutic approaches and diagnosis [31].
Sequestering miRNAs is also an important mechanism of lncRNA function in cardiac fibrosis. In the mouse MI model, lncRNA metastasis-associated lung adenocarcinoma transcript 1 (Malat1) increases sixfold when compared to controls. Loss of Malat1 attenuated the cardiac dysfunction induced by MI, resulting in the recovery of the percentages of fractional shortening (FS) and ejection fraction (EF). When MI hearts were transduced with Malat1 siRNA using lentiviral delivery, infarct size and fibrotic area were both reduced. In addition, the expression levels of collagen I and collagen III, which are fibroblast markers, were decreased. Angiotensin II (AgII) is known to be involved in both fibroblast proliferation and myofibroblast transdifferentiation in the heart. When Malat1 was decreased in AgII-treated neonatal mouse cardiac fibroblasts (NMCFs), the levels of collagen and alpha-smooth muscle actin were reduced. Conversely, Malat1 promotes cardiac fibrosis after MI by sponging miR-145. miR-145 functions in the suppression of transforming growth factor-beta1 (TGF-β1) by reducing Furin expression. In sum, Malat1 appears to cause cardiac fibrosis by limiting miR-145-dependent suppression of TGF-β1 [32].
Abnormal increases in the lncRNA myocardial infarction-associated transcript (Miat) are observed in the MI mouse model which results in cardiac interstitial fibrosis. Conversely, ectopic expression of siRNA targeting Miat in the MI mouse model restores the normal ratio of EF and FS. In addition, infarct size and fibrosis area were reduced in this model. Also, gene expression of collagen I and collagen III were reduced. An in vitro study that treated AgII in NMCF revealed that Miat activates Furin/Tgf-β1 by inhibiting miR-24 as a ceRNA, and that this resulted in cardiac fibrosis [33].
lncRNAs in cardiomyopathy
Cardiomyopathy manifests as myocardial or electrical dysfunction within the heart [34]. According to the Global Burden of Disease (GBD) 2020 study, cardiomyopathy and myocarditis caused 0.37 million deaths in 2020 and affected 6.11 million people globally during that year [14]. Eight cases out of approximately 100,000 are diagnosed with cardiomyopathy each year and classically fall into three types of cardiomyopathy: hypertropic cardiomyopathy (HCM), dilated cardiomyopathy (DCM), and restrictive cardiomyopathy (RCM).
HCM is characterized by marked myocardial hypertrophy without preexisting overload and, in about 50% of cases, it is hereditary in nature [11, 12]. For example, 30–60% of HCM patients have sarcomere variants [14]. HCM can arise at any age, but onset at young age can result in more serious disease symptoms [35]. Figure 3 shows how lncRNAs associated with HCM function.

The regulatory functions involved in cardiac hypertrophy
Some lncRNAs function as scaffolds, guides, and/or signaling molecules within the nucleus during pathogenesis. The myosin heavy chain-associated RNA transcript myosin heavy-chain-associated RNA transcripts (Mhrt) functions as cardio-protective because it inhibits the development of myocardial hypertrophy. Under pathological stress, BRG1 forms a chromatin repressor complex with histone deacetylase (HDAC) and poly-ADP ribose polymerase (PARP) proteins to suppress Mhrt expression. Alternately, Mhrt acts as a guide RNA that antagonizes the chromatin remodeling function of brahma-related gene-1 (BRG1) by binding to the BRG1 helicase domain and thereby interfering with genomic targeting of BRG1. Human Mhrt has been shown to have a conserved function as a modulator of myocardial function during cardiomyopathy. In hearts under stress, including those with hypertrophic, ischemic, or idiopathic cardiomyopathy, there was a significant decrease in Mhrt levels, with reductions of 82.8%, 72.8%, and 65.9% observed in in vitro studies of human heart. Another study showed that patients with chronic heart failure demonstrated significantly lower expression levels of Mhrt in plasma when compared to healthy individuals (p < 0.05). Additionally, individuals with higher levels of Mhrt exhibited a significantly improved overall survival rate when compared to those with lower levels of this lncRNA [36,37,38,39].
The lncRNAs antihypertropic interrelated transcript(Ahit) and cardiac-hypertropy associated factor (Chaer) function as scaffold molecules. Ahit is maximally expressed at 6 weeks in hypertrophy-induced TAC-operated mice. When Ahit was overexpressed under hypertrophic stress, increases in cell size and hypertrophic markers such as atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP), and beta-myosin heavy chain (β-MHC) were blunted. This anti-hypertrophic effect of Ahit is due to direct Ahit binding to suppressor of zeste 12 protein homolog (SUZ12), a component of the polycomb repressor complex 2 (PRC2) complex, and this event blocks the regulation of myocyte enhancer factor 2A (Mef2A) in cis. Ahit also has a human homolog, the leukemia-associated noncoding IGF1R activator RNA1 (LUNAR1) [38].
The lncRNA Chaer was found to be downregulated in the pressure overload-induced mouse model. When Chaer is diminished, hypertrophic growth of cardiomyocytes is inhibited and hypertrophic genes such as natriuretic peptide A (Nppa), β-MHC, and skeletal muscle alpha-actin (Acta1) are reduced. In contrast, overexpression of Chaer induces cardiomyocyte enlargement. After hypertrophic stimulation, the mammalian target of rapamycin (mTOR) signaling pathway-dependent lncRNA Chaer reduces H3K27 trimethylation of genes linked to pathological changes by binding to enhancer of zeste 2 polycomb repressive complex 2 subunit (EZH2), a methyltransferase subunit of PRC2. Moreover, Chaer is conserved in both rodents and humans [40].
The lncRNA cardiac hypertrophy-associated transcript (Chast) functions as a signaling molecule. Overexpression of Chast results in an increased cardiomyocyte size and expression of hypertrophic markers. Chast has a binding site for nuclear factor of activated T cells (NFAT), a pro-hypertrophic transcription factor, within its promoter region, and is upregulated by activated NFAT signaling. By suppressing pleckstrin homology and RUN domain containing M1 (PlekhM1), cardiomyocyte autophagy is prevented, and hypertrophy is induced by Chast. Chast has a homologous sequence in humans and the human form is also upregulated in patients with aortic stenosis that drives cellular hypertrophy [41].
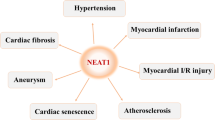
The majority of lncRNAs associated with cardiac hypertrophy function within the cytosol. The upregulation of cardiac hypertropy related factor (Chrf) is known to induce apoptosis in cardiomyocytes, and downregulation of Chrf decreases ANF and β-MHC levels, both of which are hypertrophic-related genes. Chrf also increases the myeloid differentiation primary response gene 88 (Myd88) protein by sponging the miRNA miR-489. In addition, the sequence of Chrf is not fully conserved between species, but the miR-489 binding site of Chrf is conserved between mouse and human. Further, the expression of human Chrf is increased in human heart failure [38]. The lncRNA nuclear paraspeckle assembly transcript 1 (Neat1) also functions as a sponge for miR-19a-13p and thus prevents miR-19a-13p from quenching the [Su(var)3–9, enhancer-of-zeste and trithorax] (SET) and myeloid, nervy, and DEAF-1 (MYND) domain-containing protein SET and MYND domain containing 2 (Smyd2) mRNA transcript. Through this mechanism, Neat1 upregulates Smyd2 to generate and promote cardiac hypertrophy [42].
The lncRNA phospholipid scramblase 4 (Plscr4) is expressed at a higher levels in cardiomyocytes than in cardiac fibroblasts. It is increased in TAC mice and cardiomyocytes treated with Ang II to create a hypertrophic state. Plscr4 plays a role in preventing hypertrophy and in induced hypertrophy models, depletion of Plscr4 results in an increase in the hypertrophic markers ANP, BMP, β-MHC, as well as hypertrophic cardiomyocytes. Overexpression of Plsr4 attenuates hypertrophic markers and mitigates cardiac hypertrophy in induced cardiac hypertrophy. Plscr4 acts as an endogenous sponge and sequesters miR-214 which functions in suppressing the protein mitifusion2 (Mfn2). In the presence of Plscr4, miR-214 is unable to function resulting in Mfn2 activation. Mfn2 is located in the mitochondrial outer membrane and serves as a negative regulator of cardiac hypertrophy by participating in mitochondrial fusion. Thus, lncRNA Plscr4 represses cardiac hypertrophy through the miR-214/Mfn2 axis [42]
DCM refers to cardiomyopathy characterized by systolic failure with myocardial thinning and ventricular enlargement, and most often occurs before the age of 50. Abnormal heart rhythms or heart valve abnormalities complicate the course of DCM [43, 44]. Figure 4 shows how lncRNAs associated with DCM function.

The regulatory functions involved in dilated cardiomyopathy
The lncRNA ZNF593-AS (ENST00000448923.2) has been identified in patients with DCM. Decreased expression of ZNF593-AS results in loss of cardiac contractile function because ZNF593-AS guides for heterogeneous nuclear ribonucleoproteins C1/C2 (HNRNPC) to stabilize the ryanodine receptor 2 (RYR2) mRNA transcript. RYR2 is a major component of the calcium receptor present in the sarcoplasmic reticulum and is involved in calcium handling and the contractility of cardiac muscle. Moreover, the destabilization of RYR2 by ZNF593-AS depletion results in abnormalities in cardiac contractility [45].
The lncRNA H19 is encoded by a 2.7 kb gene that is maternally expressed and paternally imprinted. It is located close to the telomeric region of chromosome 11p15.5 and is mutually imprinted and regulated with its adjacent gene, insulin-like growth factor 2 (IGF2) [42,43,44]. The expression of lncRNA H19 is increased in a mouse model of DCM,and when H19 is lost, the rate of apoptosis in cardiomyocytes is reduced and cardiac function is improved. During DCM, H19 sequesters miR-675 which, in turn, inactivates proliferation-associated 2G4 (PA2G4), a potential regulator of receptor tyrosine-protein kinase erbB-2 (ErbB3) signaling that promotes cell differentiation, apoptosis, and cell growth. In sum, the H19/miR-675/PA2G4 axis appears to significantly regulate doxorubicin-induced DCM [46].
lncRNAs in cardiac proliferation and development
So far in this manuscript we have discussed how lncRNAs function in CVD and which may potentially be therapeutic targets. We now direct our attention to lncRNAs that possess restorative capabilities from a slightly different perspective. Studies using nuclear bomb tests with carbon-14 have been performed on genomic DNA harvested from human myocardial cells. Cardiomyocytes increase by 1% per year at the age of 25, and this decreases to 0.45% by age 75. Thus, the limited ability of cardiomyocytes to regenerate markedly declines with age [47]. Cardiomyocyte loss and insufficient cardiomyocyte regeneration result in most CVDs, especially in MI. Infarction-induced heart failure results in a 25% reduction in left ventricle cardiomyocytes and leads to the death of approximately up to 1 billion myocardial cells [48]. Therefore, we investigated how lncRNAs can promote cardiomyocyte proliferation or promote pre-existing cardiomyocytes (i.e., endogenous regeneration) as a way to regenerate heart tissue. We also examined the lncRNAs involved in cardiac differentiation that enable exogenous transplantation [49].
Several lncRNAs activate, or repress, cardiomyocyte proliferation both within and outside of the nucleus. Figure 5 illustrates the mechanism by which lncRNAs activate cardiomyocyte proliferation. Examples of lncRNAs that activate cardiomyocyte proliferation in the nucleus include ECRAR, NR_045363, and Sirt1-as. The lncRNA endogenous cardiac-associated regulator (ECRAR), a signal-regulating lncRNA, is significantly upregulated in fetal heart when compared to adult heart. Ectopic expression of ECRAR in rat ventricular cardiomyocytes promoted cardiomyocyte proliferation. Also, the mitosis-specific histone mark, phosphorylated histone H3 (pH3), was increased. Although ECRAR promotes cardiac proliferation, it does not appear to induce cardiac hypertrophy. Cardiomyocyte proliferation provoked by ECRAR occurs through the extracellular signal-regulated protein kinases 1 and 2 (ERK 1/2) pathway, which plays an important role in cell cycle regulation. Positive feedback occurs when E2F transcription factor 1(E2F1), a downstream protein target of ECRAR, elevates ECRAR expression by binding within the ECRAR promoter [50].

The regulatory functions involved in activating cardiomyocyte proliferation
Other lncRNAs are involved in mRNA stabilization and miRNA binding within the cytoplasm. The lncRNA Sirt1 antisense (Sirt1-as) lncRNA, which is highly expressed in embryonic mouse hearts, overlaps with a region within the Sirt1 mRNA 3′ UTR. Overexpression of Sirt1-as has been shown to promote cardiomyocyte proliferation and increase the proliferation marker Ki67, with identical results obtained both in vivo and in vitro. When Sirt1-as was injected into an MI mouse model, it increased the animal survival rate from 56.7 to 83.3%, FS and EF, while reducing infarct size compared to controls. Sirt1-as also inhibits apoptosis and decreases cardiomyocyte size due to Sirt1-as binding, and consequential stabilization of the Sirt1transcript. Adequate levels of Sirt1 in the heart protect this organ from oxidative stress as well as attenuating apoptosis [51].
The highly conserved lncRNA NR_045363 (1700024F13Rik) is a mouse orthologue of ENST00000435695 (LOC101927497), an lncRNA that is antisense to human cyclin-dependent kinase 6 (CDK6) mRNA transcript. The expression of this lncRNA is higher in the embryonic mouse heart than in the adult heart and is specifically expressed in cardiomyocytes. In the MI model, mice expressing ectopic NR_045363 showed an increase in heart regenerative capacity compared to the control group, displayed improved EF and FS, and showed a significantly reduced infarct size. NR_045363 appears to positively control cardiomyocyte mitotic activity and proliferation by binding miR-216a and activating JAK2-STAT3 phosphorylation [52].
Representative lncRNAs that inhibit cardiomyocyte proliferation include dachshund homolog 1 (DACH1) and cardiomyocyte regeneration-related lncRNA (CRRL). The lncRNA DACH1 is a signaling molecule that is highly conserved between mouse and human, and is strikingly upregulated during postnatal heart development. lncDACH1 is increased in the hearts of MI mice compared to controls and overexpression of lncDACH1 in Myh6-lncDACH1transgenic mouse shows a decreased rate of cardiomyocyte proliferation and reduced cardiac regeneration capacity. Conversely, MI-induced mouse models in mice with cardiac-specific lncDACH1 knockout show an increased rate of cardiomyocyte proliferation, improved cardiac function, such as a higher ratio of EF and FS, and reduced infarct size compared to controls. lncDACH1 binds to inorganic pyrophosphatase 1 (PPA1) and restricts its dephosphorylation activity and also regulates yes1 associated transcriptional regulator (YAP1) signaling by increasing YAP1 phosphorylation and decreasing its nuclear translocation by binding PP1A [53].
lncRNA CRRL (ENST00000525927.5) is markedly upregulated in adult heart cardiomyocytes. In an MI model created by left anterior descending artery (LAD) ligation, loss of CRRL using RNA interference restored the ratio of left ventricular EF and left ventricular FS, and reduced infarct size. Also, depletion of CRRL in cardiomyocytes resulted in cardiomyocyte proliferation without inducing cardiac hypertrophy. In this setting, CRRL appears to act as a ceRNA by sequestering miR-119a-3p. This results in increases in the activity of a target, Hoxp1, which is known to be a key suppressor of embryonic cardiomyocyte differentiation [54]. Figure 6 depicts the mechanisms of lncRNAs that inhibit cardiomyocyte proliferation.

The regulatory functions involved in suppressing cardiomyocyte proliferation
Various lncRNAs are required for cardiac differentiation. The manner in which lncRNAs related to cardiomyocyte differentiation function is illustrated in Fig. 7. The lncRNA Braveheart (Bvht; AK143250) is located on mouse chromosome 18:61,799,307–61,807,126 (+ strand, mm9) and consists of a ~ 590 nucleotide transcript encoding three exons. It is highly expressed in mouse heart tissue and differentiation of mouse embryonic stem cells (mESCs) to cardiomyocytes shows that Bvht regulates a core network in cardiovascular development. Bvht plays an important role in the differentiation of nascent mesoderm to cardiomyocytes by inhibiting mesoderm posterior bHLH transcription factor 1 (MesP1) through interaction with SUZ12, leading to cardiac lineage commitment [55].

The regulatory functions involved in cardiomyocyte differentiation
The lncRNA Linc1405 is transcribed in close proximity to eomesodermin (eomes), a mesoderm gene, and is abundant in heart tissue of the embryo. During cardiac differentiation from mESCs, Linc1405 acts as a scaffold that drives cardiac differentiation by regulating the MesP1 gene. Exon 2 of Linc1405 binds to eomes and the Linc1405/eomes complex further complexes with WD repeat domain 5 (WDR5) and general control non-depressible 5 (GCN5) within the enhancer region of MesP1. In a mouse model, loss of Linc1405 resulted in reduced EF and FS, as well as decreased ventricular wall thickness [56].
The lncRNA modulating second heart field progenitor Moshe, 1010001N08Rik-203, is located in an antisense orientation to the GATA binding protein 6 (Gata6) gene. It is expressed in the embryonic heart at E8.5, E9.5, and E12.5, and during cardiomyocyte differentiation, Moshe suppresses cardiomyocyte differentiation by downregulating secondary heart field genes. Moshe further inactivates cardiomyocytes by binding to the NK2 homeobox 5 (Nkx2.5) promoter and Moshe is highly conserved among species [57].
Therapeutic approaches in lncRNA
lncRNAs represent auspicious therapeutic targets in CVD. Their tissue specificity and low expression levels mean that they can be treated with a limited amount of drugs with a resultant low toxicity. Using lncRNA for therapeutic purposes can be done by either removing disease-causing lncRNA or delivering disease-relieving lncRNA to the lesion site.
Therapeutic technologies using lncRNA have been developed mainly in an approach that targets their sequences [58]. siRNAs are short, double-stranded RNAs with 21 base pairs that precisely match their target lncRNA. siRNA are recognized by the RNA-induced silencing complex (RISC) and this results in target RNA degradation. The antisense strand also binds to the complementary target lncRNA leading to argonaut, the core of the RISC, performing endonucleolytic cleavage by P-element induced Wimpy testis (PIWI) domain [59].
As opposed to siRNAs, short hairpin RNA (shRNA) are synthesized in the nucleus of cells, further processed and transported into the cytoplasm, and then associate with RISC for their activity. shRNA can be expressed either transiently or stably [58, 60].
Antisense oligonucleotides (ASOs) are single-stranded RNA molecules of approximately 4 to 10 kDa in mass. ASO contain 13–25 nucleotides in their structure and also contain at least a 6-mer DNA molecule within their central domain. ASOs form ASO/RNA heteroduplexes with lncRNA via their DNA moiety and this leads to endogenous RNAse H-mediated RNA cleavage. Because ASOs do not bind to other factors such as RISC, they act not only on mature RNA transcripts but also on pre-RNA. Moreover, ASO make various chemical modifications possible that result in increased stability, binding force, and cell permeability. For example, LNA (locked nucleic acid), a nucleic acid derivative containing bicyclic furanose unit which bridges oxygen atoms on the 2′ position sugar and 4′ carbon of the ribose increases binding affinity. Other modifications include neutral backbones such as phosphorodiamidate morpholino oligomer, peptide nucleic acid, and phosphorothioates [58, 61,62,63]. These approaches take advantage of strong target specificity and provide a potentially convenient transition from the laboratory to the clinic. In addition, given current progress in interfering RNA techniques, necessary development time for therapeutic development may prove to be shortened [64].
Other approaches for targeting lncRNAs include small molecules, ribozymes, and deoxyribozymes. Conventional small molecules bind to either highly structured lncRNA or RNA-binding proteins. They alter their secondary or tertiary structure, or mask protein binding sequences within the targeted lncRNA. Therefore, in order to make “druggable” small molecules to target lncRNA, a detailed understanding of the lncRNA and binding protein structure is required [63]. Ribozymes or deoxyribozymes, such as hammerhead ribozyme, are enzymatic nucleic acid molecules that bind to complementary target sequences and catalyze RNA cleavage. These molecules cleave target lncRNAs in a site-specific manner without enzymatic proteins such as RISC or RNase H [58, 65, 66].
lncRNA delivery systems have been developed to deliver therapeutic lncRNAs or lncRNA-targeting molecules into lesions. Lentiviral vectors are the most commonly used lncRNA-carrying delivery systems as they have a packing capacity of up to 8 kb [67] and can infect both dividing and non-dividing cells. Lentiviral vectors also have a higher transduction rate and lead to non-viral protein expression after transduction. Due to integration into the genome, lentiviral vectors may provide stable long-term transgene expression [68].
Adenoviral vectors are also widely used for lncRNA overexpression and suppression. These viruses have a packing capacity of up to 5 kb and can transduce both dividing and non-dividing cells similar to lentiviral vectors. Adenovirus is a non-encapsulated, linear double-strand DNA virus [69], and therefore, no genomic integration occurs and the expression is transient in nature. Even though lentiviral vectors are not used in clinical trials, adenovirus vectors have found use in the clinic [70].
Non-viral vectors, such as liposomes, are the earliest version of lipid nanoparticle (LNP) delivery platforms. Compared with viral vectors, liposomes reduce immunogenicity and toxicity by not integrating into the genome, and can provide short-term, transient, and high-level expression of transported materials, including nucleic acids, small molecules, and proteins. Liposomes have versatile features for transporting hydrophobic and hydrophilic molecules via endocytosis, and this allows them to transduce both dividing and non-dividing cells [69, 70]. Numerous liposomal drugs have been successfully applied in clinical practice [71].
The next generation of nanoparticles (NP) consists of nucleic acid complexes with cationic lipid, nanostructured lipid carriers, peptides, polymers, and polysaccharides. These delivery systems remain solid at physiological temperatures and exhibit enhanced stability [69, 70]. Because LNPs can control the accurate location and duration of the delivered drug, they are suitable for various drug delivery applications [71].
Recently, endogenous delivery systems have been actively studied. Exosomes are extracellular vesicles (EVs) ranging in diameter from 30 to 200 nm (average ~ 100 nm), which can fuses with the cell’s plasma membrane [69]. Johnstone et al. discovered EVs in adult sheep reticulocytes in 1983. These structures were initially termed exosomes [72] and were thought to be cellular debris. Since these seminal studies, accumulating evidence shows that exosomes contain DNA, mRNA, miRNA, and lncRNA, and facilitate communication between cells [73]. They directly engage with a receptor on target cells, can transfer the receptor of the origin cell to the recipient cell, and deliver the contents of the vesicle. By this mechanism, EVs regulate cell-to-cell communication [74] and these characteristics suggest that EVs can be used for lncRNA delivery. Compared to viral or non-viral exogenous vesicles, exosomes can package drugs using patient-derived cells as the source, resulting in lower immunogenicity and higher stability. Exosomes can also be combined with liposomes, organic nanoparticles, or organic nanoparticles, such as exosome-liposome hybrids, to increase target specificity and delivery system controllability [75, 76].
Conclusions
Despite technical advances in cardiovascular medicine and well-designed emergency medical systems, CVD and especially AMI remain the leading cause of mortality and morbidity in western countries [77, 78]. Immediate reperfusion therapy can save many lives, but reperfusion often induces reperfusion injury which is thought to be mediated by increased oxygen free radicals [79,80,81,82]. Complex molecular pathway makes the issue of selecting the most appropriate therapeutic target more complicated, resulting in a lack of effective treatments to reduce reperfusion injury at the present time [83,84,85]. Similarly, many trials were attempted to regenerate cardiomyocyte by infusion or direct injection of pluripotent stem cells. Carbone et al. summarized 95 studies related to acute myocardial infarction from 2000 to 2020, categorizing those that showed benefits, as well as those that showed no effect or uncertain benefits [86,87,88,89]. Due to safety and efficacy problems, there are no stem cell therapies specifically approved by the US Food and Drug Administration (FDA) for the treatment of heart disease, even though a large number of clinics offer various cellular treatments without having gone through the FDA approval process [90]. Therefore, clinical benefit is ambiguous. It is time to renew interest in novel therapeutic targets and develop more novel approaches for better treatment of CVD. In this article, we have highlighted molecular bases of CVD in detail and suggested potential value of lncRNA as well selected molecules as therapeutic targets.
Once characterized as biological noise, lncRNA are now recognized as an important therapeutic target. Growing evidence indicates the principal roles of lncRNAs in complicated regulatory networks that govern cardiac dysfunction and regeneration. Many lncRNAs can be used as crucial therapeutic targets for CVDs, including cardiac hypertrophy, DCM, MI, and cardiac fibrosis. The concept that cardiomyocyte proliferation might be enhanced to improve cardiac regeneration led us to catalog and discuss many lncRNAs that are important in orchestrating this process. Advances in our understanding of lncRNA mechanisms involving cardiac dysfunction and cardiac regeneration underpin the potential for lncRNA therapies.
Natural molecules that target lncRNAs in specific diseases are known. For example, resveratrol targets metastasis associated lung adenocarcinoma transcript 1 (Malat1) in Parkinson’s disease [91] and noncoding nuclear-enriched abundant transcript 2 (Neat1) in myeloma [92]. Curcumin targets H19 in gastric cancer cells [93] and Malat1 in colon cancer cells [94]. Additionally, berberine targets 538 lncRNAs in non-alcoholic fatty liver disease [95]. More natural compounds targeting lncRNAs can be found on the Clinical Trial.gov website; however, it is important to note that natural compounds do not usually target lncRNAs as their principal target.
The good news is that RNA therapy is advancing. In the past few decades, significant efforts have been made to introduce RNA-based therapeutics into the clinic. As a result, several RNA-based therapeutics such as Eteplirsen, Nusinersen, and Inotersen, each of which is based on ASO technology, have been approved by the Food and Drug Administration (FDA) [96, 97]. This underscores the clinical potential of lncRNA targeted therapy.
However, in order for RNA-based therapy to be successfully applied clinically and commercialized, several limitations must be recognized and improved before drugs targeting lncRNAs can be used to treat CVDs. Even if lncRNAs expressed in human diseases are discovered, animal experiments are required before going to clinical trial, but lncRNAs are generally poorly conserved among species. In addition, even if lncRNAs are conserved in humans and mice, responses may differ between species. Another limitation is the issue of immunogenicity. As a viral defense mechanism, immune response recognizes foreign RNAs via various pathogen-associated molecular pattern (PAMP) receptors. Further, nucleic acid-based therapy can cause off-target effects. For example, sugar moiety modification can increase the probability of off-target cleavage. In fact, only 10–50% of the designed ASOs decrease the intended target [98]. The final hurdle is drug delivery. Therapeutic RNA must cross the cell membrane and, even if it passes through the cell membrane, the lncRNA must escape the endosome to enter the cytoplasm. LNPs, polymers, RNA conjugation, NPs, and virus-based approaches are under development to improve the efficacy of delivery problems. For lysosome rupture, neutrally charged ionizable lipid can be used to release nucleic acid-based drugs more easily [58, 64, 99, 100].
In spite of these difficulties, ‘third-generation gene therapy,’ which aims to treat diseases by artificially regulating gene expression at the transcription level, as exemplified by drugs like Eteplisen (Exondys 51TM), Nusinersen, and Inostersen mentioned above [96, 97], will overcome the limitations of many protein-targeted drugs. Currently, drugs targeting only mRNA and miRNA have been released, but in the near future, drugs targeting lncRNAs, especially those important in CVD pathogenesis, are expected to emerge.
Abbreviations
- ENCODE:
-
The Encyclopedia of DNA Elements
- lncRNAs:
-
Long noncoding RNAs
- ceRNA:
-
Competing endogenous RNA
- TWAS:
-
Transcriptional genome-wide studies
- CVD:
-
Cardiovascular disease
- WHO:
-
World Health Organization
- NCDs:
-
Non-communicable diseases
- NHANES:
-
National Health and Nutrition Examination Survey
- GBD:
-
Global Burden Disease
- MI:
-
Myocardial infarction
- ARIC:
-
Atherosclerosis Risk in Communities
- Snhg1:
-
Small nucleolar RNA host gene 1
- PTEN:
-
Phosphatase and tensin homolog
- PI3K:
-
Phosphatidylinositol 3′-kinase
- VEGFA:
-
Vascular endothelial growth factor A
- Phb2:
-
Prohinin2
- LDH:
-
Lactate dehydrogenase
- Oip5-as1:
-
Opa-interacting protein 5-antisense transcript 1
- Sirt1:
-
Sirtuin-1
- Ampk:
-
AMP-activated protein kinase
- Pgc1-α:
-
Peroxisome proliferator-activated receptor gamma coactivator 1-alpha
- HFrEF:
-
Heart failure with reduced ejection fraction
- HFpEF:
-
Heart failure with preserved ejection fraction
- Meg3:
-
Maternally expressed gene 3
- Mmp2:
-
Matrix metallopeptidase 2
- TAC:
-
Transverse aortic constriction
- ECM:
-
Extracellular matrix
- Wisper:
-
Wisp2 super-enhancer-associated RNA
- TIA1:
-
T-cell intracellular antigen 1
- Tiar:
-
T-cell intracellular antigen 1 (TIA1)-related/like protein
- Plod2:
-
Procollagen-lysine,2-oxoglutarate 5-dioxygenase 2
- Malat1:
-
Metastasis-associated lung adenocarcinoma transcript 1
- FS:
-
Fractional shortening
- EF:
-
Ejection fraction
- AgII:
-
Angiotensin II
- NMCFs:
-
Neonatal mouse cardiac fibroblasts
- TGF-β1:
-
Transforming growth factor-beta1
- Miat:
-
Myocardial infarction-associated transcript
- GBD:
-
Global Burden of Disease
- HCM:
-
Hypertropic cardiomyopathy
- DCM:
-
Dilated cardiomyopathy
- RCM:
-
Restrictive cardiomyopathy
- HDAC:
-
Histone deacetylase
- PARP:
-
Poly-ADP ribose polymerase
- Mhrt:
-
Myosin heavy-chain-associated RNA transcripts
- BRG1:
-
Brahma-related gene-1
- Ahit:
-
Antihypertropic interrelated transcript
- Chaer:
-
Cardiac-hypertropy associated factor
- ANP:
-
Atrial natriuretic peptide
- BNP:
-
Brain natriuretic peptide
- β-MHC:
-
Beta-myosin heavy chain
- SUZ12:
-
Suppressor of zeste 12 protein homolog
- Mef2A:
-
Myocyte enhancer factor 2A
- LUNAR1:
-
The leukemia-associated noncoding IGF1R activator RNA1
- Nppa:
-
Natriuretic peptide A
- Acta1:
-
Skeletal muscle alpha-actin
- mTOR:
-
Mammalian target of rapamycin
- EZH2:
-
Enhancer of zeste 2 polycomb repressive complex 2 subunit
- PlekhM1:
-
Pleckstrin homology and RUN domain containing M1
- Chrf:
-
Cardiac hypertropy related factor
- Neat1:
-
Nuclear paraspeckle assembly transcript 1
- MYND:
-
Myeloid, Nervy, and DEAF-1
- Smyd2:
-
SET and MYND domain containing 2
- Plscr4:
-
Phospholipid scramblase 4
- RYR2:
-
Ryanodine receptor 2
- PA2G4:
-
Proliferation-associated 2G4
- ErbB3:
-
Receptor tyrosine-protein kinase erbB-2
- ECRAR:
-
Endogenous cardiac-associated regulator
- pH3:
-
Phosphorylated histone H3
- ERK1/2:
-
Extracellular signal-regulated protein kinases 1 and 2
- E2F1:
-
E2F transcription factor 1
- Sirt1-as:
-
Sirt1 antisense
- CDK6:
-
Cyclin-dependent kinase 6
- DACH1:
-
Dachshund homolog 1
- PPA1:
-
Pyrophosphatase 1
- YAP1:
-
Yes1 associated transcriptional regulator
- MesP1:
-
Mesoderm posterior bHLH transcription factor 1
- Eomes:
-
Eomesodermin
- WDR5:
-
WD repeat domain 5
- GCN5:
-
General control non-depressible 5
- Nkx2.5:
-
NK2 homeobox 5
- RISC:
-
RNA-induced silencing complex
- PIWI:
-
P-element induced Wimpy testis
- shRNA:
-
Short hairpin RNA
- ASOs:
-
Antisense oligonucleotides
- LNA:
-
Locked nucleic acid
- LNP:
-
Lipid nanoparticle
- NP:
-
Nanoparticles
- EVs:
-
Extracellular vesicles
- MALAT 1:
-
Metastasis associated lung adenocarcinoma transcript 1
- NEAT2:
-
Noncoding nuclear-enriched abundant transcript 2
- FDA:
-
Food and Drug Administration
- PAMPs:
-
Pathogen-associated molecular patterns
References
Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG et al (2001) The sequence of the human genome. Science 291:1304–1351
Sequencing HG (2004) Finishing the euchromatic sequence of the human genome. Nature 431:931–945
Lander ES (2011) Initial impact of the sequencing of the human genome. Nature 470:187–197
Ling H, Vincent K, Pichler M, Fodde R, Berindan-Neagoe I, Slack FJ et al (2015) Junk DNA and the long non-coding RNA twist in cancer genetics. Oncogene 34:5003–5011
Lee H, Zhang Z, Krause HM (2019) Long noncoding RNAs and repetitive elements: junk or intimate evolutionary partners? Trends Genet 35:892–902
Uszczynska-Ratajczak B, Lagarde J, Frankish A, Guigó R, Johnson R (2018) Towards a complete map of the human long non-coding RNA transcriptome. Nat Rev Genet 19:535–548
Statello L, Guo C-J, Chen L-L, Huarte M (2021) Gene regulation by long non-coding RNAs and its biological functions. Nat Rev Mol Cell Biol 22:96–118
Gao N, Li Y, Li J, Gao Z, Yang Z, Li Y et al (2020) Long non-coding RNAs: the regulatory mechanisms, research strategies, and future directions in cancers. Front Oncol 10:598817
Gao L, Zhao Y, Ma X, Zhang L (2021) Integrated analysis of lncRNA–miRNA–mRNA ceRNA network and the potential prognosis indicators in sarcomas. BMC Med Genomics 14:1–11
de Goede OM, Nachun DC, Ferraro NM, Gloudemans MJ, Rao AS, Smail C et al (2021) Population-scale tissue transcriptomics maps long non-coding RNAs to complex disease. Cell 184:2633–2648. e19
Organization WH (2019) Global action plan for the prevention and control of NCDs 2013–2020. 2013. Geneva: WHO
Cheng Y, Mou L, Li Z (2022) Trends in adherence to recommended physical activity and its association with cardiovascular risk factors in US adults with cardiovascular disease: a cross-sectional study. BMC Cardiovasc Disord 22:1–7
World Health Organization. Cardiovascular diseases (CVDs) Fact sheet. https://www.who.int/en/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds). Accessed 18 Jan 2021
Tsao CW, Aday AW, Almarzooq ZI, Alonso A, Beaton AZ, Bittencourt MS et al (2022) Heart disease and stroke statistics—2022 update: a report from the American Heart Association. Circulation. 145:e153–e639
Lu L, Liu M, Sun R, Zheng Y, Zhang P (2015) Myocardial infarction: symptoms and treatments. Cell Biochem Biophys 72:865–867
Ojha N, Dhamoon AS (2021) Myocardial infarction. StatPearls [Internet]: StatPearls Publishing
Hospital NV (2020) Cardiovascular risk in men - why is heart disease a male problem. https://www.newvictoria.co.uk/about-us/news-and-articles/cardiovascular-risk-in-men-why-is-heart-disease-a-male-problem. Accessed 25 Nov 2020
Millett ER, Peters SA, Woodward M (2018) Sex differences in risk factors for myocardial infarction: cohort study of UK Biobank participants. BMJ 363:3–6
Albrektsen G, Heuch I, Løchen M-L, Thelle DS, Wilsgaard T, Njølstad I et al (2016) Lifelong gender gap in risk of incident myocardial infarction: the Tromsø study. JAMA Intern Med 176:1673–1679
Li M, Han Y, Chen Y, Li B, Chen G, Chen X et al (2021) LncRNA Snhg1-driven self-reinforcing regulatory network promoted cardiac regeneration and repair after myocardial infarction. Theranostics 11:9397
Li L, Wang J, Zhang H (2018) LncRNA-CARl in a rat model of myocardial infarction. Eur Rev Med Pharmacol Sci 22:4332–4340
Wang K, Long B, Zhou L-Y, Liu F, Zhou Q-Y, Liu C-Y et al (2014) CARL lncRNA inhibits anoxia-induced mitochondrial fission and apoptosis in cardiomyocytes by impairing miR-539-dependent PHB2 downregulation. Nat Commun 5:1–13
Mo Y, Wu H, Zheng X, Xu L, Liu L, Liu Z (2021) LncRNA CHRF aggravates myocardial ischemia/reperfusion injury by enhancing autophagy via modulation of the miR-182-5p/ATG7 pathway. J Biochem Mol Toxicol 35:e22709
Niu X, Pu S, Ling C, Xu J, Wang J, Sun S et al (2020) lncRNA Oip5-as1 attenuates myocardial ischaemia/reperfusion injury by sponging miR-29a to activate the SIRT1/AMPK/PGC1α pathway. Cell Prolif 53:e12818
Gao L, Liu Y, Guo S, Yao R, Wu L, Xiao L et al (2017) Circulating long noncoding RNA HOTAIR is an essential mediator of acute myocardial infarction. Cell Physiol Biochem 44:1497–1508
Yao J, Ma R, Wang C, Zhao G (2022) LncRNA-HOTAIR inhibits H9c2 apoptosis after acute myocardial infarction via miR-206/FN1 axis. Biochem Genet 1–12
Hinderer S, Schenke-Layland K (2019) Cardiac fibrosis–a short review of causes and therapeutic strategies. Adv Drug Deliv Rev 146:77–82
Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JG, Coats AJ et al (2016) 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Kardiologia Polska (Polish Heart Journal) 74:1037–1147
Matsusaka H, Ide T, Matsushima S, Ikeuchi M, Kubota T, Sunagawa K et al (2006) Targeted deletion of matrix metalloproteinase 2 ameliorates myocardial remodeling in mice with chronic pressure overload. Hypertension 47:711–717
Piccoli M-T, Gupta SK, Viereck J, Foinquinos A, Samolovac S, Kramer FL et al (2017) Inhibition of the cardiac fibroblast–enriched lncRNA Meg3 prevents cardiac fibrosis and diastolic dysfunction. Circ Res 121:575–583
Micheletti R, Plaisance I, Abraham BJ, Sarre A, Ting C-C, Alexanian M et al (2017) The long noncoding RNA Wisper controls cardiac fibrosis and remodeling. Sci Transl Med 9:eaai9118
Huang S, Zhang L, Song J, Wang Z, Huang X, Guo Z et al (2019) Long noncoding RNA MALAT1 mediates cardiac fibrosis in experimental postinfarct myocardium mice model. J Cell Physiol 234:2997–3006
Qu X, Du Y, Shu Y, Gao M, Sun F, Luo S et al (2017) MIAT is a pro-fibrotic long non-coding RNA governing cardiac fibrosis in post-infarct myocardium. Sci Rep 7:1–11
Brieler J, Breeden MA, Tucker J (2017) Cardiomyopathy: an overview. Am Fam Physician 96:640–646
Maron BJ, Desai MY, Nishimura RA, Spirito P, Rakowski H, Towbin JA et al (2022) Management of hypertrophic cardiomyopathy: JACC state-of-the-art review. J Am Coll Cardiol 79:390–414
Han P, Li W, Lin C-H, Yang J, Shang C, Nurnberg ST et al (2014) A long noncoding RNA protects the heart from pathological hypertrophy. Nature 514:102–106
Zhang J, Gao C, Meng M, Tang H (2016) Long noncoding RNA MHRT protects cardiomyocytes against H2O2-induced apoptosis. Biomol Ther 24:19
Forini F, Nicolini G, Kusmic C, D’Aurizio R, Mercatanti A, Iervasi G et al (2020) T3 critically affects the Mhrt/Brg1 axis to regulate the cardiac MHC switch: role of an epigenetic cross-talk. Cells 9:2155
Zhang L-E, Wu Y-J, Zhang S-L (2019) Circulating lncRNA MHRT predicts survival of patients with chronic heart failure. J Geriatr Cardiol JGC 16:818
Wang Z, Zhang X-J, Ji Y-X, Zhang P, Deng K-Q, Gong J et al (2016) The long noncoding RNA Chaer defines an epigenetic checkpoint in cardiac hypertrophy. Nat Med 22:1131–1139
Viereck J, Kumarswamy R, Foinquinos A, Xiao K, Avramopoulos P, Kunz M et al (2016) Long noncoding RNA Chast promotes cardiac remodeling. Sci Transl Med 8:326ra22-ra22
Sun X, Lv J, Dou L, Chen D, Zhu Y, Hu X (2020) LncRNA NEAT1 promotes cardiac hypertrophy through microRNA-19a-3p/SMYD2 axis. Eur Rev Med Pharmacol Sci 24:1367–1377
Andersson C, Schou M, Schwartz B, Vasan RS, Christiansen MN, D’Souza M et al (2022) Incidence rates of dilated cardiomyopathy in adult first-degree relatives versus matched controls. IJC Heart Vasc 41:101065
Hagar A, Pu X-B, Chen S-J, Shah J-P, Chen M (2019) Clinical characteristics, treatment and prognosis of patients with idiopathic dilated cardiomyopathy: a tertiary center experience. J Geriatr Cardiol JGC 16:320
Fan J, Li H, Xie R, Zhang X, Nie X, Shi X et al (2021) LncRNA ZNF593-AS alleviates contractile dysfunction in dilated cardiomyopathy. Circ Res 128:1708–1723
Zhang Y, Zhang M, Xu W, Chen J, Zhou X (2017) The long non-coding RNA H19 promotes cardiomyocyte apoptosis in dilated cardiomyopathy. Oncotarget 8:28588
Bergmann O, Bhardwaj RD, Bernard S, Zdunek S, Barnabé-Heider F, Walsh S et al (2009) Evidence for cardiomyocyte renewal in humans. Science 324:98–102
Giacca M (2020) Cardiac regeneration after myocardial infarction: an approachable goal. Curr Cardiol Rep 22:1–8
Doppler SA, Deutsch M-A, Serpooshan V, Li G, Dzilic E, Lange R et al (2017) Mammalian heart regeneration: the race to the finish line. Circ Res 120:630–632
Chen Y, Li X, Li B, Wang H, Li M, Huang S et al (2019) Long non-coding RNA ECRAR triggers post-natal myocardial regeneration by activating ERK1/2 signaling. Mol Ther 27:29–45
Li B, Hu Y, Li X, Jin G, Chen X, Chen G et al (2018) Sirt1 antisense long noncoding RNA promotes cardiomyocyte proliferation by enhancing the stability of Sirt1. J Am Heart Assoc 7:e009700
Wang J, Chen X, Shen D, Ge D, Chen J, Pei J et al (2019) A long noncoding RNA NR_045363 controls cardiomyocyte proliferation and cardiac repair. J Mol Cell Cardiol 127:105–114
Cai B, Ma W, Wang X, Sukhareva N, Hua B, Zhang L et al (2020) Targeting LncDACH1 promotes cardiac repair and regeneration after myocardium infarction. Cell Death Differ 27:2158–2175
Chen G, Li H, Li X, Li B, Zhong L, Huang S et al (2018) Loss of long non-coding RNA CRRL promotes cardiomyocyte regeneration and improves cardiac repair by functioning as a competing endogenous RNA. J Mol Cell Cardiol 122:152–164
Klattenhoff CA, Scheuermann JC, Surface LE, Bradley RK, Fields PA, Steinhauser ML et al (2013) Braveheart, a long noncoding RNA required for cardiovascular lineage commitment. Cell 152:570–583
Guo X, Xu Y, Wang Z, Wu Y, Chen J, Wang G et al (2018) A Linc1405/Eomes complex promotes cardiac mesoderm specification and cardiogenesis. Cell Stem Cell 22:893–908. e6
Kim N-J, Lee K-H, Son Y, Nam A-R, Moon E-H, Pyun J-H et al (2021) Spatiotemporal expression of long noncoding RNA Moshe modulates heart cell lineage commitment. RNA Biol 18:640–654
Fathi DB (2020) Strategies to target long non-coding RNAs in cancer treatment: progress and challenges. Egyp J Med Human Genet 21:1–15
Wilson RC, Doudna JA (2013) Molecular mechanisms of RNA interference. Annu Rev Biophys 42:217–239
Rao DD, Vorhies JS, Senzer N, Nemunaitis J (2009) siRNA vs. shRNA: similarities and differences. Adv Drug Deliv Rev 61:746–759
Ling H (2016) Non-coding RNAs: therapeutic strategies and delivery systems. Non-coding RNAs in Colorectal Cancer. Adv Exp Med Biol 937(229–237):231–233. Springer
Juliano RL (2016) The delivery of therapeutic oligonucleotides. Nucleic Acids Res 44:6518–6548
Montes M, Arnes L (2021) lncRNAs: potential therapeutic targets and biomarkers for pancreatic cancer? Expert Opin Ther Targets 25:521–528
Li X, Qi H, Cui W, Wang Z, Fu X, Li T et al (2022) Recent advances in targeted delivery of non-coding RNA-based therapeutics for atherosclerosis. Mol Ther 3121
Park JY, Lee JE, Park JB, Yoo H, Lee S-H, Kim JH (2014) Roles of long non-coding RNAs on tumorigenesis and glioma development. Brain Tumor Res Treat 2:1
Parasramka MA, Maji S, Matsuda A, Yan IK, Patel T (2016) Long non-coding RNAs as novel targets for therapy in hepatocellular carcinoma. Pharmacol Ther 161:67–78
Manjunath N, Wu H, Subramanya S, Shankar P (2009) Lentiviral delivery of short hairpin RNAs. Adv Drug Deliv Rev 61:732–745
Naldini L, Blomer U, Gallay P, Ory D, Mulligan R, Gage FH et al (1996) In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 272:263–267
Chen Y, Li Z, Chen X, Zhang S (2021) Long non-coding RNAs: from disease code to drug role. Acta Pharm Sin B 11:340–354
Bisserier M, Sun XQ, Fazal S, Turnbull IC, Bonnet S, Hadri L (2022) Novel insights into the therapeutic potential of lung-targeted gene transfer in the most common respiratory diseases. Cells 11:3–12
Tenchov R, Bird R, Curtze AE, Zhou QQ (2021) Lipid nanoparticles-from liposomes to mRNA vaccine delivery, a landscape of research diversity and advancement. ACS Nano 15:16982–17015
Johnstone RM, Adam M, Hammond JR, Orr L, Turbide C (1987) Vesicle Formation during reticulocyte maturation - association of plasma-membrane activities with released vesicles (exosomes). J Biol Chem 262:9412–9420
Han SQ, Qi YQ, Luo YM, Chen XP, Liang HF (2021) Exosomal long non-coding RNA: interaction between cancer cells and non-cancer cells. Front Oncol 10:1–7
Andaloussi EL, S, Lakhal S, Mager I, Wood MJA. (2013) Exosomes for targeted siRNA delivery across biological barriers. Adv Drug Deliv Rev 65:391–397
Tao SC, Guo SC, Zhang CQ (2018) Modularized extracellular vesicles: the dawn of prospective personalized and precision medicine. Adv Sci 5:2–9
Lin Y, Wu JH, Gu WH, Huang YL, Tong ZC, Huang LJ et al (2018) Exosome-liposome hybrid nanoparticles deliver CRISPR/Cas9 system in MSCs. Adv Sci 5
Roger VL (2007) Epidemiology of myocardial infarction. Med Clin North Am 91:537–552
Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M et al (2015) Heart disease and stroke statistics—2015 update: a report from the American Heart Association. Circulation 131:e29–e322
Reimer KA, Tanaka M, Murry CE, Richard VJ, Jennings RB (1990) Evaluation of free radical injury in myocardium. Toxicol Pathol 18:470–480
Richard V, Murry C, Jennings R, Reimer K (1990) Oxygen-derived free radicals and postischemic myocardial reperfusion: therapeutic implications. Fundam Clin Pharmacol 4:85–103
Kloner RA (1988) Introduction to the role of oxygen radicals in myocardial ischemia and infarction. Free Radical Biol Med 4:5–7
Jain A, Mohan A, Gupta O, Jajoo U, Kalantri S, Srivastava L (2000) Role of oxygen free radicals in causing endothelial damage in acute myocardial infarction. J Assoc Physicians India 48:478–480
Sharma V, Bell RM, Yellon DM (2012) Targeting reperfusion injury in acute myocardial infarction: a review of reperfusion injury pharmacotherapy. Expert Opin Pharmacother 13:1153–1175
Batchelor R, Liu DH, Bloom J, Noaman S, Chan W (2020) Association of periprocedural intravenous morphine use on clinical outcomes in ST-elevation myocardial infarction (STEMI) treated by primary percutaneous coronary intervention: Systematic review and meta-analysis. Catheter Cardiovasc Interv 96:76–88
Hoedemaker NP, Roolvink V, de Winter RJ, van Royen N, Fuster V, García-Ruiz JM et al (2020) Early intravenous beta-blockers in patients undergoing primary percutaneous coronary intervention for ST-segment elevation myocardial infarction: a patient-pooled meta-analysis of randomized clinical trials. Eur Heart J Acute Cardiovasc Care 9:469–477
Carbone R, Monselise A, Bottino G, Negrini S, Puppo F (2021) Stem cells therapy in acute myocardial infarction: a new era? Clin Exp Med 21:231–237
Mathur A, Fernández-Avilés F, Bartunek J, Belmans A, Crea F, Dowlut S et al (2020) The effect of intracoronary infusion of bone marrow-derived mononuclear cells on all-cause mortality in acute myocardial infarction: the BAMI trial. Eur Heart J 41:3702–3710
Nicolau JC, Furtado RH, Silva SA, Rochitte CE, Rassi A Jr, Moraes JB Jr et al (2018) Stem-cell therapy in ST-segment elevation myocardial infarction with reduced ejection fraction: a multicenter, double-blind randomized trial. Clin Cardiol 41:392–399
Quyyumi AA, Vasquez A, Kereiakes DJ, Klapholz M, Schaer GL, Abdel-Latif A et al (2017) PreSERVE-AMI: a randomized, double-blind, placebo-controlled clinical trial of intracoronary administration of autologous CD34+ cells in patients with left ventricular dysfunction post STEMI. Circ Res 120:324–331
Lindeman A, Pepine CJ, March KL (2020) Cardiac stem cell therapy among Clinics of Uncertain Regulatory Status (COURS): under-regulated, under-observed, incompletely understood. J Transl Med 18:1–7
Xia D, Sui R, Zhang Z (2019) Administration of resveratrol improved Parkinson’s disease-like phenotype by suppressing apoptosis of neurons via modulating the MALAT1/miR-129/SNCA signaling pathway. J Cell Biochem 120:4942–4951
Geng W, Guo X, Zhang L, Ma Y, Wang L, Liu Z et al (2018) Resveratrol inhibits proliferation, migration and invasion of multiple myeloma cells via NEAT1-mediated Wnt/β-catenin signaling pathway. Biomed Pharmacother 107:484–494
Liu G, Xiang T, Wu QF, Wang WX (2016) Curcumin suppresses the proliferation of gastric cancer cells by downregulating H19. Oncol Lett 12:5156–5162
Wei D, Yun L, Dejun X, Cong L, He J-H, Yan L (2019) Curcumin combining with si-MALAT1 inhibits the invasion and migration of colon cancer SW480 cells. Braz J Pharm Sci 55
Yuan X, Wang J, Tang X, Li Y, Xia P, Gao X (2015) Berberine ameliorates nonalcoholic fatty liver disease by a global modulation of hepatic mRNA and lncRNA expression profiles. J Transl Med 13:1–11
Stein CA, Castanotto D (2017) FDA-approved oligonucleotide therapies in 2017. Mol Ther 25:1069–1075
Rüger J, Ioannou S, Castanotto D, Stein CA (2020) Oligonucleotides to the (gene) rescue: FDA approvals 2017–2019. Trends Pharmacol Sci 41:27–41
Crooke ST (2007) Antisense drug technology: principles, strategies, and applications. CRC press
Winkle M, El-Daly SM, Fabbri M, Calin GA (2021) Noncoding RNA therapeutics—challenges and potential solutions. Nat Rev Drug Discov 20:629–651
Hueso M, Mallén A, Suñé-Pou M, Aran JM, Suñé-Negre JM, Navarro E (2021) ncRNAs in therapeutics: challenges and limitations in nucleic acid-based drug delivery. Int J Mol Sci 22:11596
Liu C-Y, Zhang Y-H, Li R-B, Zhou L-Y, An T, Zhang R-C et al (2018) LncRNA CAIF inhibits autophagy and attenuates myocardial infarction by blocking p53-mediated myocardin transcription. Nat Commun 9:1–12
Ponnusamy M, Liu F, Zhang Y-H, Li R-B, Zhai M, Liu F et al (2019) Long noncoding RNA CPR (cardiomyocyte proliferation regulator) regulates cardiomyocyte proliferation and cardiac repair. Circulation 139:2668–2684
Li X, Zhou J, Huang K (2017) Inhibition of the lncRNA Mirt1 attenuates acute myocardial infarction by suppressing NF-κB activation. Cell Physiol Biochem 42:1153–1164
Zhuang A, Calkin AC, Lau S, Kiriazis H, Donner DG, Liu Y et al (2021) Loss of the long non-coding RNA OIP5-AS1 exacerbates heart failure in a sex-specific manner. iScience 24:102537
Wang K, Liu C-Y, Zhou L-Y, Wang J-X, Wang M, Zhao B et al (2015) APF lncRNA regulates autophagy and myocardial infarction by targeting miR-188-3p. Nat Commun 6:1–11
Zeng H, Hu F, Duan Y, Li H, Wang Y (2022) Expression of lncRNA APF in peripheral blood of patients with acute myocardial infarction caused by coronary heart disease and its clinical significance. Int Heart J 63:742–748
Cai B, Ma W, Ding F, Zhang L, Huang Q, Wang X et al (2018) The long noncoding RNA CAREL controls cardiac regeneration. J Am Coll Cardiol 72:534–550
Liang H, Su X, Wu Q, Shan H, Lv L, Yu T et al (2020) LncRNA 2810403D21Rik/Mirf promotes ischemic myocardial injury by regulating autophagy through targeting Mir26a. Autophagy 16:1077–1091
Long B, Li N, Xu X-X, Li X-X, Xu X-J, Guo D et al (2018) Long noncoding RNA FTX regulates cardiomyocyte apoptosis by targeting miR-29b-1-5p and Bcl2l2. Biochem Biophys Res Commun 495:312–318
Li X, He X, Wang H, Li M, Huang S, Chen G et al (2018) Loss of AZIN2 splice variant facilitates endogenous cardiac regeneration. Cardiovasc Res 114:1642–1655
Huang L, Guo B, Liu S, Miao C, Li Y (2020) Inhibition of the LncRNA Gpr19 attenuates ischemia-reperfusion injury after acute myocardial infarction by inhibiting apoptosis and oxidative stress via the miR-324-5p/Mtfr1 axis. IUBMB life 72:373–383
Wang Q-S, Zhou J, Li X (2020) LncRNA UCA1 protects cardiomyocytes against hypoxia/reoxygenation induced apoptosis through inhibiting miR-143/MDM2/p53 axis. Genomics 112:574–580
Yu J, Yang Y, Xu Z, Lan C, Chen C, Li C et al (2020) Long noncoding RNA Ahit protects against cardiac hypertrophy through SUZ12 (suppressor of Zeste 12 protein homolog)-mediated downregulation of MEF2A (myocyte enhancer factor 2A). Circ Heart Fail 13:e006525
Viereck J, Bührke A, Foinquinos A, Chatterjee S, Kleeberger JA, Xiao K et al (2020) Targeting muscle-enriched long non-coding RNA H19 reverses pathological cardiac hypertrophy. Eur Heart J 41:3462–3474
Wang K, Liu F, Zhou L-Y, Long B, Yuan S-M, Wang Y et al (2014) The long noncoding RNA CHRF regulates cardiac hypertrophy by targeting miR-489. Circ Res 114:1377–1388
Lv L, Li T, Li X, Xu C, Liu Q, Jiang H et al (2018) The lncRNA Plscr4 controls cardiac hypertrophy by regulating miR-214. Mol Ther Nucleic Acids 10:387–397
Lai Y, He S, Ma L, Lin H, Ren B, Ma J et al (2017) HOTAIR functions as a competing endogenous RNA to regulate PTEN expression by inhibiting miR-19 in cardiac hypertrophy. Mol Cell Biochem 432:179–187
Li Y, Wang J, Sun L, Zhu S (2018) LncRNA myocardial infarction-associated transcript (MIAT) contributed to cardiac hypertrophy by regulating TLR4 via miR-93. Eur J Pharmacol 818:508–517
Zhu X, Yuan Y, Rao S, Wang P (2016) LncRNA MIAT enhances cardiac hypertrophy partly through sponging miR-150. Eur Rev Med Pharmacol Sci 20:3653–3660
Zhang Q, Wang F, Wang F, Wu N (2020) Long noncoding RNA MAGI1-IT1 regulates cardiac hypertrophy by modulating miR-302e/DKK1/Wnt/beta-catenin signaling pathway. J Cell Physiol 235:245–253
Wang Y, Cao R, Yang W, Qi B (2019) SP1-SYNE1-AS1-miR-525-5p feedback loop regulates Ang-II-induced cardiac hypertrophy. J Cell Physiol 234:14319–14329
Xiao L, Gu Y, Sun Y, Chen J, Wang X, Zhang Y et al (2019) The long noncoding RNA XIST regulates cardiac hypertrophy by targeting miR-101. J Cell Physiol 234:13680–13692
Trembinski DJ, Bink DI, Theodorou K, Sommer J, Fischer A, van Bergen A et al (2020) Aging-regulated anti-apoptotic long non-coding RNA Sarrah augments recovery from acute myocardial infarction. Nat Commun 11:1–14
Anderson KM, Anderson DM, McAnally JR, Shelton JM, Bassel-Duby R, Olson EN (2016) Transcription of the non-coding RNA upperhand controls Hand2 expression and heart development. Nature 539:433–436
Funding
This research was supported by grants from the Ministry of Science and ICT and the National Research Foundation of Korea (NRF) SRC program: Comparative Medicine Disease Research Center (CDRC) (2021R1A5A1033157) and National Research Foundation of Korea (NRF) (2014M3A9D5A01073598).
Author information
Authors and Affiliations
Contributions
N. Kim conducted investigations; contributed to the conception and design of the study, acquisition, and analysis; and wrote the initial version of the manuscript, and edited it. W.Y. Chung provided interpretation of reference data, analysis, review, and editing the manuscript. J.Y. Cho provided funding acquisition, conception and design of the study, supervision, review, and editing of the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Corresponding author
Ethics declarations
Ethical approval
Not applicable.
Competing interests
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kim, N., Chung, WY. & Cho, JY. The role and medical prospects of long non-coding RNAs in cardiovascular disease. Heart Fail Rev 28, 1437–1453 (2023). https://doi.org/10.1007/s10741-023-10342-1
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10741-023-10342-1