
Abstract
Heavy metals are serious inorganic pollutants that need to be monitored in the hydrosphere with simple and cheap methods. Herein, a new sensor was fabricated by modifying a carbon paste electrode with MgO/Fe2O3 nanocomposite for simple, rapid, accurate, and highly sensitive simultaneous determination of Cd (II) and Cu (II) using differential pulse anodic stripping voltammetry. The electrochemical behavior of the constructed sensor was examined, and all parameters were optimized including deposition potential, time, pH, and scan rate. For Cd (II) and Cu (II), the respective detection limits were determined to be 3.3 × 10−11 M and 3.6 × 10−11 M, and the respective quantification limits were 1.1 × 10−10 M and 1.2 × 10−10 M. The sensor estimated Cd (II) and Cu (II) in Nile river, tap, and bottled real water samples with high recoveries ranging from 99 to 117%.
Graphical Abstract

Similar content being viewed by others

Introduction
Heavy metal ions (HMIs) are the main environmental pollutant due to their toxicity and inability to biodegrade. HMIs contribute to many physiological disorders and accumulate in the body as a result of exposure affecting the immune, digestive, and nervous systems even in small doses. They are usually found in cosmetics industries, fertilizers, and other chemicals prominent from industrial or household waste [1, 2]. Hence, it is crucial to accomplish accurate and rapid identification of heavy metal ions like Cd (II) and Cu (II) [3]. In aquatic systems, cadmium (Cd) is one of the most harmful contaminants. It is difficult to remove from water systems through self-purification and could be dangerous to both the environment and people’s health [4]. Serious bone problems like osteoporosis can result from the long-term accumulation of Cd in the food chain [4]. Copper is a crucial component of both plant and animal nutrition. Moreover, because it is the third-most abundant trace element in biological systems, it is essential to numerous metabolic activities. Furthermore, copper deficiency results in anemia of a microcytic type [5]. An excessive amount of Cu (II) can cause hemochromatosis and gastrointestinal catarrh disorders as well as disrupt the balance of reactive oxygen species [1, 6].
For the determination of Cd (II) and Cu (II), a variety of techniques have been used, including atomic absorption [7], atomic fluorescence [8], inductively coupled plasma [9], and mass spectrometry [10]. Despite this, these techniques are costly, time-consuming, and difficult to perfect [6]. Electroanalytical methods, on the other hand, are efficient in detecting Cd (II) and Cu (II) and are gaining popularity due to their simplicity, high sensitivity, good selectivity, and low cost [11, 12].
Among heterogeneous carbon electrodes, carbon paste electrodes (CPEs) are a unique group [13]. CPEs are represented by carbon paste, which is a mixture of carbon (graphite) powder and an appropriate liquid binder packed into an electrode body with the proper design [13]. These electrodes are widely used mainly for voltammetric measurements due to its many advantages such as low cost, simple preparation, and ease of surface modification with a variety of modifiers [14].
In recent years, there has been a lot of interest in using metal oxide nanoparticles in sensing applications. These nanoparticles have excellent physical and catalytic properties such as their high adsorption capacity, large surface area, good mass transport, and catalytic capability, resulting in electrocatalytic performance enhancement and lower detection limits [15,16,17]. Among them, magnesium oxide MgO and hematite Fe2O3 have recently gained popularity in bio-electrochemical and electrochemical processes to improve the electrode surface in batteries and ease the transfer of electrons in various types of sensors, because they are thermodynamically stable, biocompatible, environmentally friendly, low cost, and have the ability to facilitate electron transmission [18, 19]. Fe2O3 NPs are widely applied in a variety of industrial processes due to their electrical, magnetic, and optical features [20]. Thus, Fe2O3 NPs were applied to modify CPE to determine dopamine and tramadol [21, 22]. The MgO NPs are useful in a variety of industries including electronics, coatings, ceramics, catalysis, petrochemicals, superconducting materials, paints, and antiseptics [23]. As a result, they were employed as a modifier for carbon paste electrodes in the determination of many analytes such as paracetamol [21, 23], catechol and hydroquinone [16], ezetimibe [18], desmedipham [24], and loperamide [25]. However, neither Fe2O3 NPs nor MgO NPs have been previously used to modify CPE for the detection of either Cd (II) or Cu (II) ions.
Generally, single metal oxide nanoparticles may suffer from aggregation, low surface adsorption of analyte, low selectivity and conductivity, and may have no sensitivity to specific HMIs. This limits their electrochemical applicability in the real world. In such cases, bi-metal oxide nanocomposites are developed to overcome this problem by combining the qualities of many materials to get the desired result [26]. Less aggregation of nanoparticles, higher analyte adsorption, higher catalytic activity, better catalytic selection, and higher resistance to deactivation are a few advantages that bimetallic nanomaterials have over identical monometallic ones. Thus, metal oxide nanocomposites have gained popularity recently in a variety of electrochemical technologies, particularly biosensors and bioelectronics [27]. Recently, a Bi2O3/MgO/Fe2O3 trimetallic nanocomposite has been synthesized by solution combustion method and applied for photcatalytic degradation of fast orange red dye and detection of paracetamol [28].
The most common methods for preparing mixed metal oxides are coprecipitation and thermal decomposition, which generate large surface areas more easily and cleanly and disperse homogenous particle sizes well. The prepared compounds show higher catalytic activity due to this large surface area [29].
Herein, we prepared MgO/Fe2O3 bimetallic nanocomposite using coprecipitation method and applied it to modify CPE. The new MgO/Fe2O3/CPE sensor is firstly applied for simultaneous determination of Cd (II) and Cu (II) using differential pulse anodic stripping voltammetry (DPASV) technique. The sensor achieved very low detection limits, good sensitivity, and better anti-interference ability.
Experimental
Chemicals and Reagents
All chemicals are of analytical grade and are used exactly as supplied and purchased from Merck. The preparation of a stock solution of 1 × 10−5 M of Cd (II) and Cu (II) included dissolving the required amount of each in bidistilled water, followed by the preparation of 0.1 M potassium chloride, phosphate buffer (PBS), Britton-Robinson buffer (BRB), and acetate buffer (ABS). A sodium acetate buffer was prepared daily (ADWIC, 98% purity), which was used as a supporting electrolyte. To regulate the pH values, solutions of 1.0 M HCl and 1.0 M NaOH were used. All studies were conducted at room temperature (25 °C).
Instrumentation
The EG&G Princeton Applied Research POTENTIOSTAT /GALVA NOSTAT (model 263, USA) was used as the instrument for the electrochemical experiments (chronoamperometry, cyclic voltammetry, linear sweep voltammetry, and differential pulse voltammetry) which were connected to an external computer. The electrochemical cell (model k0264 micro-cell) consists of three electrodes: Ag/AgCl-saturated KCl serves as the reference electrode (model k0265). The working electrode is an electrochemically modified CPE, whereas the counter electrode (model k0266) is made of a platinum wire of great purity. A Cyber Scan pH 500 Meter (Euteoh, India) was used to adjust the pH values.
Synthesis of MgO/Fe2O3 Nanocomposite
MgO/Fe2O3 nanocomposite was synthesized by coprecipitation method, where 6.4 g magnesium nitrate and 10.1 g iron nitrate were dissolved in 50 ml bidistilled water and stirred, heated for 5 min (80 °C), then NH4OH (1 M) was added in portions, and the reaction continued further for 30 min when the corresponding hydroxides were completely precipitated (Scheme 1) [30]. The resulted precipitate was isolated through centrifugation at 6000 rpm for 15 min, followed by washing with bidistilled H2O several times and drying at 80 °C overnight. The product was crushed using pestle and mortar and stored in a sealed container after being ignited at 500 °C for 4 h.

Synthesis of MgO/Fe2O3 nanocomposite
Preparation of Real Samples
Nile river water was collected from the surface of Nile river (Qena, Egypt), and bottled drinking water was obtained from the local market. River, bottled drinking, and tap water were filtrated with a membrane filter paper, then poured into ABS. The amounts of Cd (II) and Cu (II) in the samples were measured using the standard addition method.
Fabrication of MgO/Fe2O3/CPE
Upon preliminary experiments, in a mortar, 60% graphite powder, 15% MgO/Fe2O3 nanocomposite and 25% paraffin wax were combined for 15 min to create a homogenous paste that was then packed into an insulin syringe to create the modified electrode. The copper wire was then dipped in the mixture and used to make contact with the cell.
Results and Discussion
Fabrication of MgO/Fe2O3/CPE
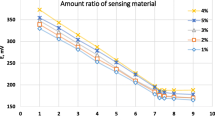
The optimum amount of MgO/Fe2O3 nanocomposite has been tested to achieve the ideal conditions for the determination of Cd (II) and Cu (II) ions. The results are shown in Fig. 1 that indicates highest current was obtained after modification of the CPE with 15% MgO/Fe2O3. Thus, it was chosen for the fabrication of MgO/Fe2O3/CPE sensor.

The amount of MgO/Fe2O3 nanocomposite in carbon paste for detection of Cd (II) and Cu (II)
XRD, FT-IR, and SEM Analysis of MgO/Fe2O3 Nanocomposite
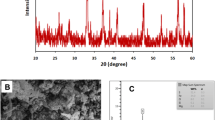
Powder X-ray diffraction (XRD) studies were used to determine the crystalline structure and average crystalline size of the synthesized MgO/Fe2O3 nanocomposite. Figure 2 displays the powder XRD patterns of the MgO/Fe2O3 nanocomposite with a respective ratio of 22.2:77.6. The sample displayed a series of diffraction peaks corresponding to the well-crystalline structure of MgO/Fe2O3 nanocomposite (COD 9009782 and COD 1011326). Using the Scherrer formula, the MgO/Fe2O3 nanocomposite has a crystallite size of approximately 41.5 nm.

XRD patterns of the MgO/Fe2O3 nanocomposite
Figure 3 reveals the FT-IR spectrum of the prepared MgO/Fe2O3 nanocomposite, which was recorded between wave numbers 4000 and 400 cm−1. The peak at 418 cm−1 is associated with Mg-O bond vibrations, while two absorption peaks are observed at 549 and 863 cm−1, which are connected to Fe–O [15]. Absorption peaks observed at 1155, 1462, and 1747 cm−1 correspond to the M-OH, Mg-O stretching, and O–H bending, respectively. The absorption peak at 3384 cm−1 is assigned to the stretching vibration of H2O molecules [30].

FT-IR spectrum of the prepared MgO/Fe2O3 nanocomposite
By using SEM technique, the surface morphology and size of the synthesized MgO/Fe2O3 nanocomposite were examined, revealing that they are nanosized and spherical in shape (Fig. 4).

SEM of the prepared MgO/Fe2O3 nanocomposite
Electrochemical Characterization
The cyclic voltammetry of the [Fe(CN)6]3−/4− redox couple were used to evaluate the MgO/Fe2O3/CPE. The cyclic voltammograms (CVs) of 2 × 10−3 M [Fe(CN)6]3−/4− in 0.1 M KCl at a scan rate 100 mV/s at bare CPE and MgO/Fe2O3/CPE are shown in Fig. 5. Small redox peak currents were seen at the bare CPE with a peak separation (ΔEpeak) of 180 mV (curve a), while well-defined oxidation and reduction peaks were seen when MgO/Fe2O3/CPE was utilized and (ΔEpeak) was obviously reduced to 82 mV. This is because of the better electron transfer through MgO/Fe2O3 between the analyte and the CPE. This confirmed that the electrode surface property was significantly changed and showed the high electrocatalytic activity of the MgO/Fe2O3/CPE towards [Fe(CN)6]3−/4− redox couple [31].

CVs of 2.0 mM [Fe(CN)6]3−/4− in 0.1 M KCl at a bare CPE (a) and the MgO/Fe2O3/CPE (b) at a scan rate 100 mV/s
Parameters Optimization for Electrochemical Detection
Experimental parameters such as deposition potential, deposition time, and supporting electrolyte were optimized. The deposition potential and deposition time are crucial variables that regulate the effectiveness of stripping linear sweep voltammetry (SLSV) analysis. The influence of the deposition potential for Cd (II) was examined between −0.9 and −1.4 V and is shown in Fig. 6a. It can be observed that the voltammetric peak current increases with increasing deposition potential from −0.9 to −1.2 V after which it decreases. Therefore, −1.2 V is selected for the subsequent experiments [32]. Also, Fig. 6b illustrates studying the deposition potential for Cu (II) between −0.9 and −1.4 V. When the deposition potential increases from −0.9 to −1.1 V, the peak current increases, and by increasing the potential over −1.1V, the response of current is reduced. From this, it is considered to choose −1.1 V as the deposition potential [3].

SLSVs of the deposition potential of 5 × 10−6 M Cd (II) in 0.1 M acetate buffer (pH 4.5) (a) and 5.2 × 10−6 M Cu (II) in 0.1 M acetate buffer (pH 5.0) (b) at scan rate 50 mV/s at MgO/Fe2O3/CPE and the deposition time of 5 × 10−6 M Cd (II) in 0.1 M acetate buffer (pH 4.5) and 5.2 × 10−6 M Cu (II) in 0.1 M acetate buffer (pH 5.0) at scan rate 50 mV/s at MgO/Fe2O3/CPE, respectively (c and d)
The deposition time and Cd (II) oxidation peak current were also investigated. The peak current increased with increasing the deposition time from 50 to 120 s due to the increase in the amount of analyte that accumulated on the electrode surface with an increase in deposition time. However, when increasing the deposition time above 120 s, the response of current decreased due to the saturation of the surface electrode with Cd ions. Hence, the best deposition time in the subsequent studies was determined to be 120 s (Fig. 6c) [33]. Figure 6d shows the impact of the deposition time between 60 and 200 s on the oxidative peak current of Cu (II). The figure clearly shows that the peak current increases as the deposition time increases from 60 to 150 s. The voltammetric peak current is reduced when applying the deposition time beyond 150 s. This may be due to the competition of H+ ions with Cu (II); therefore, 150 s was voted as the deposition time for the subsequent tests [3, 34].
The effect of several supporting electrolytes such as phosphate buffer, BRB, KCl, and acetate buffer solution on the peak currents of Cd (II) and Cu (II) were examined. The 0.1 M acetate buffer solution was chosen as the supporting electrolyte because it produced higher Cd (II) and Cu (II) peak currents with well-defined shapes as depicted in Fig. 7a and b, respectively.

SLSVs of a 5 × 10−6 M Cd (II) and b 5.2 × 10−6 M Cu (II) at MgO/Fe2O3/CPE in different supporting electrolytes
Voltammetric Responses of the Cd (II) and Cu (II) at MgO/Fe2O3/CPE
Figure 8a reveals the SLSVs of 5 × 10−6 M Cd (II) in 0.1 M acetate buffer (pH 4.5) at a scan rate 50 mV/s at bare CPE, MgO/Fe2O3/CPE, and CPE without Cd (II) and MgO/Fe2O3. As seen in Fig. 8a, no obvious peak was observed at CPE without MgO/Fe2O3 or Cd (II) (curve A), while a slight obvious response can be observed at the bare CPE (curve B). A well-defined irreversible oxidation peak at −0.73 V is shown at MgO/Fe2O3/CPE (curve C), which is displaced to negative direction [32]. A significant peak current for Cd (II) was seen in comparison with bare CPE due to the large surface area and higher catalytic activity of the mixed metal oxides [29].

a SLSVs of 0.1 M acetate buffer (pH 4.5) at the bare CPE without Cd (II) (curve A), at the bare CPE with 5 × 10−6 M Cd (II) (curve B), and at the MgO/Fe2O3/CPE (curve C) with a scan rate 50 mV/s and b SLSVs of 0.1 M acetate buffer (pH 5.0) at a scan rate 50 mV/s at the bare CPE without Cu (II) (curve A), at the bare CPE with 5.2 × 10−6 M Cu (II) (curve B), and at MgO/Fe2O3/CPE (curve C)
Also, the SLSVs of 5.2 × 10−6 M Cu (II) in 0.1 M acetate buffer (pH 5.0) at scan rate 50 mV/s are displayed in Fig. 8b for bare CPE, MgO/Fe2O3/CPE, and bare CPE without Cu (II) and MgO/Fe2O3. As can be observed, the bare CPE without Cu (II) and MgO/Fe2O3 (curve A) showed no distinct peak, but the bare CPE depicted a small oxidation peak (curve B). In MgO/Fe2O3/CPE, a well-defined, irreversible oxidation peak at −0.15 V is displayed (curve C). This peak is moved in the negative direction [32]. Moreover, there was a sharp rise in the current response which was related to the excellent electrochemical performance of MgO/Fe2O3 [35].
This demonstrates that the new MgO/Fe2O3/CPE sensor has a good catalytic performance for the oxidation of Cd (II) and Cu (II).
Effect of pH
The influence of 5 × 10−6 M Cd (II) in 0.1 M acetate buffer at a scan rate 50 mV/s and various pH values ranging from 2.5 to 7.6 was shown in Fig. 9a. The stripping current of Cd (II) increased with increasing the pH value from 2.5 to 4.5 due to more H+ ions present in the solution, which could compete with the heavy metal ions for the active sites on the surface of the electrode [36]. However, at higher pH, the peak current decreased owing to the hydrolysis of Cd (II) [32, 37], and the direction of the peak potential became negative. Therefore, pH 4.5 is the optimal. As seen in the inset of Fig. 9a, the relation between peak potential and pH is linear using the equation:

a SLSVs of MgO/Fe2O3/CPE in 0.1 M acetate buffer with pH range (2.5–7.6) for 5 × 10−6 M Cd (II) at a scan rate 50 mV/s and b pH range (2.0–7.0) for 5.2 × 10−6 M Cu (II) at a scan rate 50 mV/s. Insets: Plots of EP vs. pH for Cd (II) and Cu (II)
The slope is extremely close to the Nernstian value (0.059 V/pH at 25 °C), demonstrating a protons to electrons ratio of 1. Moreover, Fig. 9b examines the SLSVs of 5.2 × 10−6 M Cu (II) in 0.1 M acetate buffer at a scan rate 50 mV/s at different pH values from 2 to 7. By increasing the pH value from 2 to 5, the response of current increased. The anodic peak current reduced at higher pH values due to the hydrolysis of Cu (II) [38], and the peak potential shifted to the negative direction. Hence, pH 5.0 is derived as the optimum value for electrochemical testing. Peak potential and pH have a linear relationship, as shown in the inset of Fig. 9b by using the following equation:
As the slope is so near to the Nernstian value (0.059 V/pH at 25 °C), the protons/electrons ratio is 1.
Effect of Scan Rate
The effect of various scan rates on the peak current for 5 × 10−6 M Cd (II) in 0.1 M acetate buffer (pH 4.5) from 10 to 150 mV/s was evaluated in Fig. 10a. It was observed that the anodic peak current increased when the scan rate raised due to the irreversible electrooxidation reaction of Cd (II); the peak potential is moved to the positive side. A linear relationship between the peak current (Ip) of Cd (II) and the square root of the scan rate (v1/2) is shown in the inset of Fig. 10a for the same range. The resulting equation appears as follows:

a SLSVs of 5 × 10.−6 M Cd (II) in 0.1 M acetate buffer solution (pH 4.5) at the MgO/Fe2O3/CPE with various scan rates (10, 20, 50, 80, 100, and 150 mV/s) (inset: corresponding calibration curve), b calibration plot of log (Ip) and log (v), and c calibration plot of Ep versus log (v)
This implies that the reaction was diffusion-controlled at our modified electrode surface [39]. A straight line with a slope of 0.48 resulted from the plot of log Ip versus log v (Fig. 10b), which is consistent with the theoretical value of 0.5 suggesting diffusion-controlled process [39]:
A linear calibration plot of Ep versus log v using the following equation can be seen in Fig. 10c as well:
The Laviron equation was used to calculate the total number of participating electrons ΔE/Δlog (v) = 0.059/nα, (where α is the electron transfer coefficient) which was determined to be 2.
Also, Fig. 11a shows the evaluation of the impact of different scan rates on the peak current for 5.2 × 10−6 M Cu (II) in 0.1 M acetate buffer (pH 5.0) from 10 to 100 mV/s. The response of current was shown to increase as the scan rate increased. The peak potential is shifted to the positive side as a result of irreversible electrooxidation process of Cu (II). Inset of Fig. 11a over the same range illustrates a linear relationship between the peak current (Ip) of Cd (II) and the square root of the scan rate (v1/2). Following is the equation that results:

a SLSVs of 5.2 × 10−6 M Cu (II) in 0.1 M acetate buffer solution (pH 5.0) at the MgO/Fe2O3/CPE with various scan rates (10, 20, 30, 50, 80, and 100 mV/s) (inset: corresponding calibration curve), b calibration plot of log (Ip) and log (v), and c calibration plot of Ep versus log (v)
This suggests that the electron transfer reaction at modified electrode was diffusion-controlled [39]. A diffusion-controlled process was indicated by the straight line of the log (Ip) against log v with a slope of 0.48 (Fig. 11b), which is very near to the theoretical value of 0.5 [40]:
Figure 11c additionally displays a calibration plot of Ep versus log v that is linear and employs the equation:
The total number of participating electrons was determined using the Laviron equation, ΔE/Δlog (v) = 0.059/nα, where α is the electron transfer coefficient, and it was found to be 2.
Analytical Performance of the MgO/Fe2O3/CPE
In Fig. 12a, the voltammetric response of MgO/Fe2O3/CPE at various concentrations of Cd (II) in the range of 0.0001 to 5 µM was investigated using differential pulse anodic stripping voltammetry method (DPASV) under the ideal conditions. Peak current increased linearly with increasing Cd (II) concentration. It was shown that there is a linear relationship between the anodic peak current and Cd (II) concentration (inset Fig. 12a). The corresponding linear equation is:

DPASVs of the MgO/Fe2O3/CPE (a) in 0.1 M acetate buffer (pH 4.5) at deposition potential −1.2 and deposition time 120 s containing various concentrations of Cd (II) (0.0001, 0.001, 0.002, 0.01, 0.09, 0.6, 0.8, 0.9, 1.8, 2.1, 2.3, 2.6, 3.1, 3.6, 4, 4.3, and 5 µM), inset: calibration curve and b in 0.1 M acetate buffer (pH 5.0) at deposition potential −1.1and deposition time 150 s including different concentrations of Cu (II) (0.0001, 0.002, 0.006, 0.08, 0.099, 0.89, 0.99, 1.7, 2.1, 2.5, 2.9, 3.2, 3.6,4,4.4.4.8, and 5.2 µM), inset: calibration curve at pulse height 25 × 10−3 V, pulse width 50 × 10−3 s, step time 0.1 s and scan rate 20 mV/s
The following equations [34] were used to determine LOD and LOQ in this study: LOD = 3 s/m and LOQ = 10 s/m.
Where s is the standard deviation of the peak current (three runs) of the lowest concentration, and m is the slope of the corresponding calibration equation; the LOD and LOQ were determined to be 3.3 × 10−11 M and 1.1 × 10−10 M, respectively.
Similarly, DPASV was used to examine the voltammetric response of MgO/Fe2O3/CPE at different Cu (II) concentrations in the range of 0.0001 to 5.2 µM at optimum conditions, and the results are shown in Fig. 12b. Increasing Cu (II) concentration resulted in gradual increase in the peak current. Inset of Fig. 12b shows the linear relation between the anodic peak current and Cu (II) concentrations. The associated linear equation is:
The LOD and LOQ were found to be 3.6 × 10−11 M and 1.2 × 10−10 M, respectively.
Comparison of the LOD results of our constructed electrode with that of other reported modified electrodes are presented in Table 1.
Simultaneous Determination of Cd (II) and Cu (II) at MgO/Fe2O3/CPE
As shown in Fig. 13, DPASVs were used to determine various concentrations of Cd (II) and Cu (II) in 0.1 M acetate buffer solution (pH 4.7) at MgO/Fe2O3/CPE. The peak currents of the metal ions increased linearly when the concentrations of both Cd (II) and Cu (II) increased simultaneously, demonstrating the ability of MgO/Fe2O3/CPE to identify both metal ions. The obtained linear relationships with the corresponding regression equations (insets of Fig. 13) are:

DPASVs of the MgO/Fe2O3/CPE in 0.1 M acetate buffer (pH 4.7) containing various concentrations of Cd (II) (0.0009, 0.4, 0.8, 2.1, 3.1, 4.1, and 5 µM) and Cu (II) (0.0001, 0.006, 0.7, 2.2, 3.2, 4.4, and 5.2 µM) at scan rate 20 mV/s, step time 0.1 s, pulse height 25 mV, and pulse width 50 ms
LOD values are 7.3 × 10−11 M and 7.6 × 10−11 M for Cd (II) and Cu (II), respectively.
Repeatability and Stability Tests
To assess the repeatability of the modified electrode, the MgO/Fe2O3/CPE was subjected to six consecutive measurements for 5 × 10−6 M Cd (II) as shown in Fig. 14a. The response of current remained constant with relative standard deviation of 0.104%. Also, as shown in Fig. 14b, the repeatability of 5.2 × 10−6 M Cu (II) at MgO/Fe2O3/CPE was evaluated by six repetitive measurements, and the peak current remained unchanged. The relative standard deviation is 0.131%. This demonstrates the high repeatability of the MgO/Fe2O3/CPE.

a and b DPASVs of six successive measurements of 5 × 10−6 M Cd (II) and 5.2 × 10−6 M Cu (II) in 0.1 M acetate buffer using CPE modified by MgO/Fe2O3, and c, d the stability of the electrode for Cd (II) and Cu (II), respectively
The stability of the modified electrode was evaluated for 5 × 10−6 M Cd (II) and 5.2 × 10−6 M Cu (II) after keeping it at ambient temperature for 60 days. The results are shown in Fig. 14c and d. The electrode initial responses were retained by 97.1% and 97% for Cd (II) and Cu (II), respectively, confirming the high stability of the MgO/Fe2O3/CPE.
Effect of Interferences
To evaluate the selectivity of the MgO/Fe2O3/CPE, various interfering species were investigated in 0.1 M acetate buffer including 5 × 10−6 M Cd (II) and 5.2 × 10−6 M Cu (II) on the oxidation peaks of them (Table 2). The presence of 400-fold concentration ratios of the interfering ions such as Ni2+, Mg2+, Zn2+, and Pb2+. Also, 200-fold of Co2+, Ca2+, and Fe3+ and 100-fold of Na+ and K+. There is no interference effect on the estimation of Cd (II) and Cu (II) and the change peaks at less than 5%. So, even at high concentrations of these interferences, the MgO/Fe2O3/CPE is extremely selective for the detection of Cd (II) and Cu (II).
Real Samples Analysis
Using the standard addition method, MgO/Fe2O3/CPE was used to determine Cd (II) and Cu (II) in river, tap, and drinking water samples. DPASV analyses were performed after diluting the samples with 0.1 M acetate buffer. The recovery values of the spiked samples ranged between 99 and 117% for both species as shown in Table 3, indicating that the senor is highly applicable.
Conclusions
MgO/Fe2O3 nanocomposite was successfully synthesized and used to modify CPE for the simultaneous determination of Cd (II) and Cu (II). Due to the large surface area and higher catalytic activity, the modified sensor displayed excellent sensitivity, low detection limit, good stability, and great anti-interference ability for estimation of (II) and Cu (II). Real water samples can be simply, selectively, and accurately analyzed for cadmium and copper ions using the new sensor.
Availability of Data and Materials
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
J. Wang, P. Yu, K. Kan, H. Lv, Z. Liu, B. Sun, K. Shi, Efficient ultra-trace electrochemical detection of Cd2+, Pb2+ and Hg2+ based on hierarchical porous S-doped C3N4 tube bundles/graphene nanosheets composite. Chem. Eng. J. 420, 130317 (2021)
T.M. Lima, P.I. Soares, L.A. do Nascimento, D.L. Franco, A.C. Pereira, L.F. Ferreira, A novel electrochemical sensor for simultaneous determination of cadmium and lead using graphite electrodes modified with poly (p-coumaric acid). Microchem. J. 168, 106406 (2021)
D. Huo, Y. Zhang, N. Li, W. Ma, H. Liu, G. Xu, C. Hou, Three-dimensional graphene/amino-functionalized metal-organic framework for simultaneous electrochemical detection of Cd (II), Pb (II), Cu (II), and Hg (II). Anal. Bioanal. Chem. 414, 1575–1586 (2022)
N.M. Thanh, N. Van Hop, N.D. Luyen, N.H. Phong, T.T. Tam Toan, Simultaneous determination of Zn (II), Cd (II), Pb (II), and Cu (II) using differential pulse anodic stripping voltammetry at a bismuth film-modified electrode. Adv. Mater. Sci. Eng. 1–11 (2019)
E. Bilici, Z. Yazicigil, M. Tok, Y. Oztekin, Electrochemical determination of copper (II) using modified glassy carbon electrodes. Desalin. Water Treat. 50(1–3), 198–205 (2012)
B. Niu, B. Yao, M. Zhu, H. Guo, S. Ying, Z. Chen, Carbon paste electrode modified with fern leave-like MIL-47 (as) for electrochemical simultaneous detection of Pb (II), Cu (II) and Hg (II). J. Electroanal. Chem. 886, 115121 (2021)
M. Ghaedi, F. Ahmadi, A. Shokrollahi, Simultaneous preconcentration and determination of copper, nickel, cobalt and lead ions content by flame atomic absorption spectrometry. J. Hazard. Mater. 142(1–2), 272–278 (2007)
L.A. Hutton, G.D. O’Neil, T.L. Read, Z.J. Ayres, M.E. Newton, J.V. Macpherson, Electrochemical X-ray fluorescence spectroscopy for trace heavy metal analysis: enhancing X-ray fluorescence detection capabilities by four orders of magnitude. Anal. chem. 86(9), 4566–4572 (2014)
N.R. Biata, K.M. Dimpe, J. Ramontja, N. Mketo, P.N. Nomngongo, Determination of thallium in water samples using inductively coupled plasma optical emission spectrometry (ICP-OES) after ultrasonic assisted-dispersive solid phase microextraction. Microchem. J. 137, 214–222 (2018)
L. Suo, X. Dong, X. Gao, J. Xu, Z. Huang, J. Ye, L. Zhao, Silica-coated magnetic graphene oxide nanocomposite based magnetic solid phase extraction of trace amounts of heavy metals in water samples prior to determination by inductively coupled plasma mass spectrometry. Microchem. J. 149, 104039 (2019)
W. Ye, Y. Li, J. Wang, B. Li, Y. Cui, Y. Yang, G. Qian, Electrochemical detection of trace heavy metal ions using a Ln-MOF modified glass carbon electrode. J. Solid State Chem. 281, 121032 (2020)
W. Zhang, S. Fan, X. Li, S. Liu, D. Duan, L. Leng, L. Qu, Electrochemical determination of lead (II) and copper (II) by using phytic acid and polypyrrole functionalized metal-organic frameworks. Microchim. Acta 187, 1–9 (2020)
I. Švancara, K. Vytřas, K. Kalcher, A. Walcarius, J. Wang, Carbon paste electrodes in facts, numbers, and notes: a review on the occasion of the 50‐years jubilee of carbon paste in electrochemistry and electroanalysis. Electroynalysis (N.Y.N.Y.). 21(1), 7–28 (2009)
R.F. Aglan, G.G. Mohamed, H.A. Mohamed, Determination of aluminum (III) by using a modified carbon paste selective electrode. J. Pharm. Res. 5(9), 4748–4754 (2012)
A.K. Ns, S. Ashoka, M. Pandurangappa, MgFe2O4 nanoparticles synthesis and characterization: application to trace level mercury (II) measurement from waste water samples. Mater. Res. Express. 6(12), 125049 (2019)
K. Chetankumar, B.K. Swamy, T.S.K. Naik, A reliable electrochemical sensor for detection of catechol and hydroquinone at MgO/GO modified carbon paste electrode. J. Mater. Sci. Mater. Electron. 31, 19728–19740 (2020)
N. Mohammadizadeh, S.Z. Mohammadi, M. Kaykhaii, Carbon paste electrode modified with ZrO2 nanoparticles and ionic liquid for sensing of dopamine in the presence of uric acid. J. Anal. Chem. 73, 685–694 (2018)
S. Chitravathi, S. Reddy, B.K. Swamy, Electrochemical determination of ezetimibe by MgO nanoflakes-modified carbon paste electrode. J. Electroanal. Chem. 764, 1–6 (2016)
T. Alizadeh, F. Zargr, Highly selective and sensitive iodide sensor based on carbon paste electrode modified with nanosized sulfate-doped α-Fe2O3. Mater. Chem. Phys. 240, 122118 (2020)
M.M. Vinay, Y.A. Nayaka, Iron oxide (Fe2O3) nanoparticles modified carbon paste electrode as an advanced material for electrochemical investigation of paracetamol and dopamine. J. Sci. Adv. Mater. Dev. 4(3), 442–450 (2019)
M. Irdhawati, N.K.Y.S. Manurung, Lestari, Preparation and validation of Fe203 modified carbon paste electrode for measurement of dopamine by the voltammetry method. Jurnal Kimia Sains dan Aplikasi 22(6), 227–234 (2019)
A. Vazirirad, A. Babaei, M. Afrasiabi, SnO2/α-Fe2O3 Hierarchical nanorods modified carbon paste electrode as the novel sensor for sensitive simultaneous determination of dopamine and tramadol. Anal. Bioanal. Electrochem. 13(3), 393–407 (2021)
K.G. Manjunatha, B.K. Swamy, H.D. Madhuchandra, K.J. Gururaj, K.A. Vishnumurthy, Synthesis and characterization of MgO nanoparticle and their surfactant modified carbon paste electrode sensing for paracetamol. Sens. 2, 100127 (2021)
E. Demir, A simple and sensitive square wave stripping pathway for the analysis of desmedipham herbicide by modified carbon paste electrode based on hematite (α-Fe2O3 nanoparticles). Electroanalysis 31(8), 1545–1553 (2019)
N. Al-Qasmi, M.T. Soomro, M. Aslam, A.U. Rehman, S. Ali, E.Y. Danish, A. Hameed, The efficacy of the ZnO: α-Fe2O3 composites modified carbon paste electrode for the sensitive electrochemical detection of loperamide: a detailed investigation. J. Electroanal. Chem. 783, 112–124 (2016)
H. Huang, L. Chen, S. Wang, P. Kang, X. Chen, Z. Guo, X. J. Huang, Electrochemical monitoring of persistent toxic substances using metal oxide and its composite nanomaterials: design, preparation, and application. TrAC, Trends Anal. Chem. 119, 115636 (2019)
P. Kannan, G. Maduraiveeran, Metal oxides nanomaterials and nanocomposite-based electrochemical sensors for healthcare applications. Biosens. 13(5), 542 (2023)
H. Madanakumara, H.S. Jayanna, C.V. Yelamaggad, S. Soundeswaran, M. Vishwas, K.S. Shamala, B.S. Surendra, N. Basavaraju, Enhanced electrochemical sensor and photodegradation of industrial wastewater by Almond gum assisted synthesis of Bi2O3/MgO/Fe2O3 nanocomposites. Sensors International 3, 100193 (2022)
E. Mosaddegh, A. Hassankhani, Preparation, characterization, and catalytic activity of Ca2CuO3/CaCu2O3/CaO nanocomposite as a novel and bio-derived mixed metal oxide catalyst in the green synthesis of 2H-indazolo [2, 1-b] phthalazine-triones. Catal. Commun. 71, 65–69 (2015)
S.A. El-Molla, H.R. Mahmoud, Synthesis, textural and catalytic properties of nanosized Fe2O3/MgO system. Mater. Res. Bull. 48(10), 4105–4111 (2013)
M.F. Mousavi, M. Amiri, A. Noori, S.M. Khoshfetrat, A prostate specific antigen immunosensor based on biotinylated-antibody/cyclodextrin inclusion complex: fabrication and electrochemical studies. Electroanal. 29, 2818–2831 (2017)
R. Huang, K. Zhang, H. Sun, D. Zhang, J. Zhu, S. Zhou, G. Hu, Star-shaped porous nitrogen-doped metal-organic framework carbon as an electrochemical platform for sensitive determination of Cd (II) in environmental and tobacco samples. Anal. Chim. Acta 1228, 340309 (2022)
S. Deshmukh, G. Kandasamy, R.K. Upadhyay, G. Bhattacharya, D. Banerjee, D. Maity, S.S. Roy, Terephthalic acid capped iron oxide nanoparticles for sensitive electrochemical detection of heavy metal ions in water. J. Electroanal. Chem. 788, 91–98 (2017)
R. Tahaei, H. Shayani-Jam, M.R. Yaftian, Voltammetric determination of trace copper (II), cadmium (II), and lead (II) using a Schiff base modified glassy carbon working electrode. Monatsh. Für. Chem. 152, 51–59 (2021)
M. Ognjanović, D.M. Stanković, M. Fabián, A. Vukadinović, Z. Prijović, B. Dojčinović, B. Antić, A voltammetric sensor based on MgFe2O4 decorated on reduced graphene oxide-modified electrode for sensitive and simultaneous determination of Catechol and Hydroquinone. Electroanal. 30(11), 2620–2627 (2018)
Y. Zhang, C. Li, Y. Su, W. Mu, X. Han, Simultaneous detection of trace Cd (II) and Pb (II) by differential pulse anodic stripping voltammetry using a bismuth oxycarbide/nafion electrode. Inorg. Chem. Commun. 111, 107672 (2020)
L. Zhu, L. Xu, B. Huang, N. Jia, L. Tan, S. Yao, Simultaneous determination of Cd (II) and Pb (II) using square wave anodic stripping voltammetry at a gold nanoparticle-graphene-cysteine composite modified bismuth film electrode. Electrochim. Acta 115, 471–477 (2014)
Z. Guo, X.K. Luo, Y.H. Li, Q.N. Zhao, M.M. Li, Y.T. Zhao, C. Ma, Simultaneous determination of trace Cd (II), Pb (II) and Cu (II) by differential pulse anodic stripping voltammetry using a reduced graphene oxide-chitosan/poly-l-lysine nanocomposite modified glassy carbon electrode. J. colloid interface sci. 490, 11–22 (2017)
F. Khaleghi, Z. Arab, V.K. Gupta, M.R. Ganjali, P. Norouzi, N. Atar, M.L. Yola, Fabrication of novel electrochemical sensor for determination of ascorbic acid in the presence of folic acid in food and pharmaceutical samples. J. Mol. Liq. 221, 666–672 (2016)
M.R. Ganjali, F.G. Nejad, H. Beitollahi, S. Jahani, M. Rezapour, B. Larijani, Highly sensitive voltammetric sensor for determination of ascorbic acid using graphite screen printed electrode modified with ZnO/Al2O3 nanocomposite. Int. J. Electrochem. Sci. 12(4), 3231–3240 (2017)
J. Zhou, K. Pan, G. Qu, W. Ji, P. Ning, H. Tang, R. Xie, rGO/MWCNTs-COOH 3D hybrid network as a high-performance electrochemical sensing platform of screen-printed carbon electrodes with an ultra-wide detection range of Cd (II) and Pb (II). Chem. Eng. J. 449, 137853 (2022)
L.L. Fernández, J. Bastos-Arrieta, C. Palet, M. Baeza, Composite electrodes based on carbon materials decorated with hg nanoparticles for the simultaneous detection of Cd (II), Pb (II) and Cu (II). Chemosensors. 10(4), 148 (2022)
M. Ramalingam, V.K. Ponnusamy, S.N. Sangilimuthu, A nanocomposite consisting of porous graphitic carbon nitride nanosheets and oxidized multiwalled carbon nanotubes for simultaneous stripping voltammetric determination of cadmium (II), mercury (II), lead (II) and zinc (II). Microchim. Acta 186, 1–10 (2019)
N. Mohamad Nor, S. Arivalakan, N.D. Zakaria, N. Nilamani, Z. Lockman, K. Abdul Razak, Self-assembled iron oxide nanoparticle-modified APTES-ITO electrode for simultaneous stripping analysis of Cd (II) and Pb (II) ions. ACS omega. 7(4), 3823–3833 (2022)
Z. Xu, Z. Liu, M. Xiao, L. Jiang, C. Yi, A smartphone-based quantitative point-of-care testing (POCT) system for simultaneous detection of multiple heavy metal ions. Chem. Eng. J. 394, 124966 (2020)
K.M. Hassan, G.M. Elhaddad, M. AbdelAzzem, Voltammetric determination of cadmium (II), lead (II) and copper (II) with a glassy carbon electrode modified with silver nanoparticles deposited on poly (1, 8-diaminonaphthalene). Microchim. Acta 186, 1–10 (2019)
X. Zheng, S. Chen, J. Chen, Y. Guo, J. Penga X. Zhou, R. Lv, J. Lina, R. Lin, Highly sensitive determination of lead(II) and cadmium(II) by a large surface area mesoporous alumina modified carbon paste electrode. RSC Adv. 8, 7883 (2018).
M. Malakootian, H. Abolghasemi, H. Mahmoudi-Moghaddam, A novel electrochemical sensor based on the modified carbon paste using Eu3+ − doped NiO for simultaneous determination of Pb (II) and Cd (II) in food samples. J. Electroanal. Chem. 876, 114474 (2020)
L. Liu, C. Yu, X. Zhang, H. Ma, C. Ma, Y. Fu, X. Dong, Carbon paste electrode modified with bismuth oxychloride as a sensor for the determination of Pb2+ and Cd2+. Int. J. Electrochem. Sci. 14, 4469–4482 (2019)
H. Xing, J. Xu, X. Zhu, X. Duan, L. Lu, W. Wang, T. Yang, Highly sensitive simultaneous determination of cadmium (II), lead (II), copper (II), and mercury (II) ions on N-doped graphene modified electrode. J. Electroanal. Chem. 760, 52–58 (2016)
T. Zhang, H. Jin, Y. Fang, J. Guan, S. Ma, Y. Pan, M. Du, Detection of trace Cd2+, Pb2+ and Cu2+ ions via porous activated carbon supported palladium nanoparticles modified electrodes using SWASV. Mater. Chem. Phys. 225, 433–442 (2019)
S. Di Masi, A. Garcia Cruz, F. Canfarotta, T. Cowen, P. Marote, C. Malitesta, S.A. Piletsky, Synthesis and application of ion‐imprinted nanoparticles in electrochemical sensors for copper (II) determination. Chem. Nano. Mat. 5(6), 754–760 (2019).
E. Tüzün, G. Atun, Individual and simultaneous determination of heavy metal ions using carbon paste electrode modified with titania nanoparticles. Electrocatalysis (2023). https://doi.org/10.1007/s12678-023-00824-z
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB).
Author information
Authors and Affiliations
Contributions
Keriman M. Abd-Elsabur: draft manuscript preparation, experimental, analysis and interpretation of results, and software acquisition. Mohamed Abd-Elsabour: draft manuscript preparation, experimental, analysis and interpretation of results, and software acquisition. Fawzy H. Assaf: writing—review and editing. Ibrahem M.A. Hasan: draft manuscript preparation, experimental, analysis and interpretation of results, and software acquisition.
Corresponding authors
Ethics declarations
Ethical Approval
This is not applicable.
Competing Interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Abd-Elsabur, K.M., Abd-Elsabour, M., Assaf, F.H. et al. Electrochemical Estimation of Cd and Cu Ions Simultaneously Using a Modified MgO/Fe2O3 Nanocomposite/Carbon Paste Electrode. Electrocatalysis 14, 875–890 (2023). https://doi.org/10.1007/s12678-023-00843-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12678-023-00843-w