
Abstract
The interplay between crystal–melt and grain boundary interfaces in partially melted polycrystalline aggregates controls many physical properties of mantle rocks. To understand this process at the fundamental level requires improved knowledge about the interfacial structures and energetics. Here, we report the results of first-principles molecular dynamics simulations of two grain boundaries of (0l1)/[100] type for tilt angles of 30.4° and 49.6° and the corresponding solid–liquid interfaces in Mg2SiO4 forsterite at the conditions of the upper mantle. Our analysis of the simulated position time series shows that structural distortions at the solid–liquid interfacial region are stronger than intergranular interfacial distortions. The calculated formation enthalpy of the solid–solid interfaces increases nearly linearly from 1.0 to 1.4 J/m2 for the 30.4° tilt and from 0.8 to 1.0 J/m2 for the 49.6° tilt with pressure from 0 to 16 GPa at 1500 K, being consistent with the experimental data. The solid–liquid interfacial enthalpy takes comparable values in the range 0.9 to 1.5 J/m2 over similar pressure interval. The dihedral angle of the forsterite–melt system estimated using these interfacial enthalpies takes values in the range of 67° to 146°, showing a decreasing trend with pressure. The predicted dihedral angle is found to be generally larger than the measured data for silicate systems, probably caused by compositional differences between the simulation and the measurements.
Similar content being viewed by others


Introduction
Grain boundaries and crystal–melt interfaces are common in melt-bearing rocks of the Earth’s interior. These interfaces control the thermodynamics and kinetics of the overall system thereby influencing many physical properties, such as elasticity, electrical conductivity, element partitioning, and permeability. For example, the grain boundary interfacial regions serve as reservoirs for incompatible elements and may facilitate ion and electron transport (Hiraga et al. 2004; Karki et al. 2015; Marquardt and Faul 2018; Pommier et al. 2018). The combined role of solid–solid and solid–liquid interfaces determines whether the liquid phase gets trapped at grain junctions or it wets the solid surface and percolates through the solid matrix (e.g., Waff and Bulau 1979; Hier-Majumdar 2006; Holness 2010; Zhimulev et al. 2018). A relevant microstructural parameter is the dihedral angle which is the angle subtended between two solid–liquid interfaces at the corner of a fluid-filled pore (Smith 1948; Holness 2006). This angle helps us determine the geometry, interconnectivity, and critical volume fraction of the liquid phase in partially melted polycrystalline aggregate.
Numerous experimental studies have investigated the role of 3- and 4-grain junctions and microstructures in geologically relevant crystal–melt/glass systems (see a review by Holness 2010). Complete wetting of forsterite and olivine grains by dry and hydrous melts was implied based on their measured small dihedral angles (< 60°) at zero and elevated pressures (Toramaru and Fujii 1986; Walker et al. 1988; Cmiral et al. 1998; Mibe et al. 1998a; Maumus et al 2004; Yoshino et al. 2007). Small dihedral angles were generally found for silicic melt-bearing rocks (Jurewicz and Watson 1985; Gleason et al. 1999). However, the Fe–C/S melts do not wet silicates resulting in dihedral angles higher than 60° (e.g., Terasaki et al. 2005; Dong et al. 2019). Computational studies of crystal–melt interfaces particularly using the first-principles approach are still rare. The grain boundaries were previously simulated in some mantle materials including Mg2SiO4 forsterite using both empirical and first-principles molecular dynamics methods (de Leeuw et al. 2000; Adjaoud et al. 2012; Ghosh and Karki 2014; Ghosh et al. 2022). These simulations investigated the structure and energetics of grain boundaries and the incorporation of defects and impurities at the interfacial regions.
Here, we report the results of large-scale first-principles molecular dynamics simulations of selected grain boundaries and crystal–melt interfaces in Mg2SiO4 forsterite—a major mantle material at the pressure conditions of the upper mantle. In particular, two (0l1)/[100] grain boundaries with tilt angles of 30.4° and 49.6° which were previously predicted to be energetically most favorable at 0 K (Adjaoud et al. 2012; Ghosh and Karki 2014; Ghosh et al. 2022) were simulated at the elevated temperature of 1500 K which is comparable to the upper mantle geotherm. Correspondingly, two solid–liquid systems using the crystal part from respective grain boundaries and amorphous configuration for the melt part were simulated for gaining insight on to the behavior of partially melted mantle rocks. Both the crystal and liquid parts of the simulated system have the same composition of Mg2SiO4 (with silica molar fraction of 0.33). The composition of the coexisting liquid is generally expected to be different from Mg2SiO4 because the melting of forsterite is incongruent at pressures above 9 GPa (e.g., Presnall and Walker 1993; Belmonte et al. 2017). Eutectic melting of the MgO–SiO2 binary system also results in different melts with silica fraction in the range of 0.4 to 0.5 (e.g., Kato and Kumazawa 1985; Presnall et al. 1998; de Koker et al. 2013). Nevertheless, our results and analysis of the iso-composition systems focus on understanding the similarities and differences in the structure and energetics of the solid–solid and solid–liquid interfaces as a function of pressure up to 16 GPa. Using the calculated interfacial enthalpies, we estimate the dihedral angle at liquid–solid–solid junction and discuss its implication for partial melts in the mantle.
Methodology
For solid–solid interface, we consider the (011)/[100] and (021)/[100] grain boundaries with respective tilt angles of 30.4° and 49.6° which were predicted to be energetically most favorable among many grain boundaries simulated at 0 K by previous first-principles studies (Ghosh and Karki 2014; Ghosh et al. 2022). We model the grain boundary system as a bi-crystal which comprises two oppositely oriented blocks (one block being the mirror image of the other block) of the crystal merged at the interface (Fig. 1). The resulting supercell thus consists of two grain–grain interfaces, one at the center and the other as split across the edges. The solid–liquid (crystal–melt) interface is generated by joining a tilted crystal region (on the left) with an amorphous region (on the right) of equal dimensions resulting in a two-phase supercell (Fig. 1). Corresponding to each tilt angle (30.4° or 49.6°) considered for the grain boundary, the tilted crystal part is cut in such a fashion that it maintains the periodicity along all crystalline directions. The amorphous part of the supercell is disordered because it uses Mg2SiO4 liquid configuration. Since the tilted configurations are cut along a particular plane, the atomic orientations of the exposed surfaces vary with the tilt angle. As in the case of the bi-crystal supercell, there are two solid–liquid interfaces, one near the center of the supercell and the other as split across the edges of the supercell. To minimize interactions between the two interfaces, the supercell is constructed in a rectangular shape with a much longer dimension in the direction normal to the interface (Fig. 1).

The structural snapshots of the simulated bi-crystal (top) and two-phase (bottom) systems for the 30.4° tilt at 10.4 and 10.2 GPa, respectively, at 1500 K. Each crystal region is an ordered arrangement of SiOn units displayed as cyan tetrahedra and MgOn units shown as bonds between Mg atoms (green spheres) and O atoms (red spheres). The molten configuration (the right half of the two-phase supercell) also contains five- and six-fold Si–O coordination species displayed as blue pentahedra and pink octahedra, respectively, thus representing a random spatial distribution of these structural units. Also drawn are the rotated orthorhombic unit cell of forsterite in each crystalline region and the tilt angle made with interfacial plane (the vertical dashed line)
First-principles simulations of these interface-bearing Mg2SiO4 systems were performed using the generalized gradient approximation (Perdew et al. 1996) and the projector augmented wave method (Kresse and Joubert 1999) as implemented in VASP (Kresse and Furthmuller 1996). Plane wave basis set with an energy cut-off of 900 eV, Gamma point Brillouin zone sampling, and an energy convergence criterion of 10–4 eV were used. The elongated rectangular supercells comprised 112 formula units (total 784 atoms) for the tilt 30.4° systems and 144 formula units (total 1008 atoms) for the tilt 49.6° systems. The supercells consisting of 32 and 56 formula units were, respectively, used to simulate bulk Mg2SiO4 in its crystalline (forsterite) and molten phases (Fig S1). For the solid–solid system, we started with static simulations corresponding to 0 K performed at different volumes with optimized cell parameters to capture the effect of pressure in the desired range. Thereafter, each of these optimized configurations was heated to 1500 K and further equilibrated for 10–20 picoseconds with a time step of 1 femtosecond depending on the convergence of energy with time. For the two-phase system, bulk crystal and melt configurations corresponding to 1500 K were used. A few simulations were also performed at 1200 and 1800 K. All simulations used NVT ensembles in which the number of atoms, volume, and temperature are constant. The internal energy (E) and pressure (P) as the direct outputs of the simulations allow us to evaluate the enthalpy \(H=E+PV\) of each simulated system and hence the interfacial enthalpy of the solid–solid and solid–liquid systems. Both energy and pressure fluctuate with time, however, their values averaged over successive time windows converge within the simulation durations (Fig. S2). Two ways of evaluating the interfacial free energy are the thermodynamic integration/cleaving wall approach and the fluctuation method (Laird and Davidchack 2005; Benet et al. 2015). However, these methods generally require much larger supercells than those used here and at the present they are too intensive computationally for the sizes of these models with our first-principles approach.
Results and analysis
Structural properties
To gain structural insights on the simulated crystal-crystal and crystal–melt systems (Fig. 1 and Fig S3), we compute partial radial distribution functions (RDFs) and local coordination environments using the atom position time series. At 1500 K, thermally induced vibrations lead to non-uniqueness in the interatomic distances in a crystal, resulting in finite width of the RDF peaks which become vertical lines at 0 K. The Si/Mg–O RDFs of the left and right crystalline parts of the bi-crystal grain boundary system are the same as expected (Fig. 2, left). These functions show a clear first peak followed by smaller peaks at larger distances, so the overall long-range order is maintained in either crystalline part. In the case of the solid–liquid system, these RDFs drastically differ between the left and right parts (Fig. 2, right). The Si–O function of the molten part shows a similar first peak as the crystalline part, but its second peak is weak and broad, apparently replacing the crystalline second and third peaks. This means that the second and third nearest oxygen neighbors of silicon atoms are mixed up in the molten part, a characteristic loss of long-range order of the crystal lattice. The Mg–O function also shows large differences between the solid and liquid parts of the two-phase system. The first peak is broader and noticeably shifted to smaller distance relative to the crystalline peak.

The Si–O and Mg–O radial distribution functions (RDF) for the simulated solid–solid (SS) and solid–liquid (SL) Mg2SiO4 systems corresponding to the 30.4° and 49.6° tilt configurations around 10 GPa and 1500 K. The RDF curves for the left and right halves of each system are shown by solid and dashed curves, respectively
It is important to note here that at relatively high temperatures, the radial distribution functions alone are insufficient to characterize the structure. By considering all Si/Mg–O pair distances which fall within the first peak of the corresponding RDF, we examine local bonding/coordination environments. The crystal part of both grain boundary and solid–liquid systems is composed of Si–O tetrahedra representing four oxygen-coordinated silicon atoms in an ordered arrangement at all pressures (Fig. 1). However, some under- and over-coordinated species appear in the vicinity of the interfacial plane for both systems. The liquid part is also composed of randomly distributed Si–O tetrahedra at low pressure. As pressure increases, the Si–O coordination distribution of Mg2SiO4 liquid picks up the pentahedral and octahedral states in significant proportions (de Koker et al 2008). The presence of long-range order in the crystalline part and its apparent absence in the liquid part are visibly clear from the visualization snapshots (Fig. 1). These differing structural characteristics of the left and right parts confirm that the supercell maintains its adjoined crystalline–amorphous regions at 1500 K.
To further enhance our structural analysis, we quenched the 1500 K configurations (Fig S3) down to 0 K so that the thermal effects are completely removed. The structural parameters for the bulk parts of the grain boundary system almost match with their crystalline counterparts. For instance, the average Si–O bond length is 1.655 Å at ~ 0 GPa and 1.635 Å at ~ 15 GPa and the Si–O coordination number is 4 for all cases (pure crystal, 30.4° and 49.6° tilt interface systems). Similarly, the Mg–O bond lengths for the bulk parts remain very close to each other and on average take slightly higher values (2.151, 2.113 Å at 0 GPa and 2.030, 2.045 Å at 15 GPa) when compared to the crystal. It is noted that in forsterite Mg has two distinct octahedral sites, one with longer and the other with shorter average bond lengths. In case of the solid–liquid system, the structural parameters of the crystalline half are like those of pure crystal and their values in the molten half are like those of the amorphous phase.
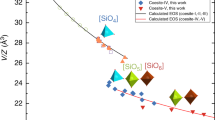
We explore local structural characteristics of the simulated systems at 0 K by evaluating the bond distortion index defined for SiOn and MgOn polyhedral units with \({\delta }_{\lambda }= \frac{1}{n}{\sum }_{i=1}^{n}\frac{{\lambda }_{i} - {\lambda }_{av}}{{\lambda }_{av}}\), where \({\lambda }_{i}\) and \({\lambda }_{av}\) are the individual and average bond lengths, respectively (Fig. 3). The index takes non-zero values for Si/Mg–O units in forsterite, so these units are not ideal polyhedra. In both crystal parts of the grain boundary system, the distortion index remains comparable to that for the bulk crystal except at the interfacial region. Around the interface plane the distortion index takes higher values for both polyhedral units. This means that the interfacial region is more distorted, and the degree of distortion is weakly sensitive to pressure.

Spatial distribution of Si–O and Mg–O distortion indices along a distance perpendicular to the interfacial plane of the quenched 0 K solid–solid (SS, circles) and solid–liquid (SL, diamonds) interfaces in Mg2SiO4 for the 30.4° and 49.6° tilt configurations around 10 GPa. Horizontal lines represent the Si/Mg–O distortion factors of the forsterite crystal
The distortion index for the Si–O bonds in the crystal part of the two-phase system closely follows the pure crystal trend, but it differs noticeably in the case of Mg–O bonds. On the other hand, both Si–O and Mg–O polyhedral units are highly distorted in the molten/amorphous part and the disorder nature seems to be fully retained covering the entire amorphous half of the system (Fig. 3). From the amorphous region to the crystal half, the distortions get diminished yet being much stronger than in the intergranular interfacial region. The liquid disorder thus seems to have penetrated the other crystal half to some extent. In overall, the crystal half of the solid–liquid supercell appears to be more distorted than either crystal half of the grain boundary supercell (Fig. 3).
Interfacial energetics
We evaluate the interfacial energy as the formation enthalpy of the solid–solid (\({\gamma }_{\text{SS}}\)) and solid–liquid (\({\gamma }_{\text{SL}}\)) interfaces (Fig. 4) by taking appropriate differences between the calculated enthalpies of the forsterite, Mg2SiO4 liquid, bi-crystal, and crystal–melt supercells divided by the interfacial surface area (Tables S1 and S2). Given that the models of the two types of interfaces considered are conceptually similar, their formation enthalpy is given by
where the subscript “int” represents solid–solid (SS) or solid–liquid (SL). Hint represents the enthalpy of the simulated bi-crystal or two-phase system and Aint is the area of the corresponding interface plane. The factor 0.5 accounts for the two interfaces contained in the supercell because of the periodic boundary conditions imposed on the supercell. Hbulk is the enthalpy of the corresponding bulk phase, which contains the same number of crystalline Mg2SiO4 units as together present in the bi-crystal system (Table S1). For the solid–liquid system the bulk phase corresponds to the sum of the numbers of crystalline and amorphous Mg2SiO4 units present in the supercell (Table S2).

Calculated interfacial enthalpy of solid–solid (SS) and solid–liquid (SL) Mg2SiO4 systems as a function of pressure at 1500 K for the 30.4° configuration (upper panel) and 49.6° configuration (lower panel). The upper panel also shows the SL interfacial enthalpy results at 1200 and 1800 K by the dashed blue and dotted red lines, respectively. The enthalpy results at 0 K are shown for the solid–solid interfaces (grain boundaries)
The 49.6° tilt grain boundary has lower interfacial enthalpy of ~ 0.6 J/m2 as compared to ~ 0.9 J/m2 for the 30.4° tilt grain boundary at zero pressure and 0 K. Both interfacial enthalpies increase almost linearly with pressure to their respective values of 1.0 and 1.4 J/m2 around 15 GPa. The calculated values of \({\gamma }_{\text{SS}}\) as a function of pressure at 0 K are consistent with the previous computations (Ghosh and Karki 2014; Ghosh et al. 2022). At 1500 K, \({\gamma }_{\text{SS}}\) also shows a linear trend with pressure with its values closely following the 0 K results (Fig. 4). It is remarkable that temperature does not influence the solid–solid interfacial enthalpy significantly. This makes sense because the bulk parts of the bi-crystal system remain mostly unchanged at 1500 K and any thermal contributions to the enthalpy for these parts are compensated by equivalent contributions from the perfect crystal. However, the interfacial region which is considerably distorted at 0 K has the possibility of additional local structural rearrangement at the elevated temperature, but these thermally induced changes are small as well. The calculated interfacial enthalpy of two tilt grain boundaries in forsterite takes values in the range 0.8 to 1.4 J/m2 between 0 and 16 GPa at 1500 K. These results are generally consistent with the experimentally inferred interfacial free energy values of 0.9 J/m2 for the olivine–basalt system (Cooper and Kohlstedt 1982) and 1.4 J/m2 for natural olivine (Duyster and Stöckhert 2001).
In the grain boundary simulation, the bulk of each crystalline part is not expected to change much, and therefore, it becomes relatively easy to deal with the SS interfacial energetics. In the case of two-phase system, while one half is crystalline, the other half is amorphous/molten. Continuous relaxation of the amorphous part although slow is unavoidable, and the relaxation time depends on temperature. The simulations at 1500 K were run for 10–20 picoseconds, so the short-time structural rearrangements (local relaxations) are likely to have occurred within that duration. As shown in Fig. 4, the solid–liquid interfacial enthalpy (\({\gamma }_{\text{SL}}\)) corresponding to the 30.4° is smaller than \({\gamma }_{\text{SS}}\) at most pressures. The 49.6° tilt case shows an opposite trend, that is, \({\gamma }_{\text{SL}}\) is larger than \({\gamma }_{\text{SS}}\) at all pressures. We also find that \({\gamma }_{\text{SL}}\) corresponding to the 30.4° tilt is lower than that for the 49.6° tilt. This can be associated with the observation that the 30.4° SL system shows weaker distortion at the interfacial region as well as in the crystal region than the 49.6° SL system (as reflected by the Mg–O bond distortion factor in Fig. 3). In contrast, \({\gamma }_{\text{SS}}\) corresponding to the 30.4° tilt is higher than that for the 49.6° tilt. In overall, the calculated \({\gamma }_{\text{SL}}\) results are considerably scattered taking values between 0.9 and 1.5 J/m2 at pressures ~ 0 to ~ 16 GPa and 1500 K and they are generally comparable with the calculated \({\gamma }_{\text{SS}}\) values. We also find the calculated values of \({\gamma }_{\text{SL}}\) at 1200 and 1800 K to be close to those at 1500 K for the 30.4° configuration (Fig. 4).
Discussion
Solid–liquid interfaces together with grain boundaries play vital role in the wetting behavior of rocks by melts. Partial melting is thought to occur in the deep mantle, and the distribution and amounts of these melts, however, remain mostly unconstrained. Whether co-existing melt with solid percolates at the interfacial region of two grains or not depends on the geometry of two-phase aggregates. The interplay between grain–melt (solid–liquid) and grain–grain (solid–solid) interfaces is manifested in the dihedral angle (θ) of liquid phase in solid matrix. For an isotropic system, this angle formed by the interfaces at the liquid–solid–solid junction (Smith 1948; Holness 2010) satisfies the following relation:
where γSS and γSL are the solid–solid and solid–liquid interfacial energies, respectively. The calculated energetics of the bi-crystal and crystal–melt systems allows us to evaluate the dihedral angle for Mg2SiO4 using Eq. 2. We consider a linear pressure variation of \({\gamma }_{\text{SS}}\) for each tilt grain boundary simulated here and use the calculated values of \({\gamma }_{\text{SL}}\) at different pressures as obtained from the simulations for the corresponding solid–liquid system as shown in Fig. 4 (Table S2). We note here that for the purpose of this study, we use the calculated interfacial enthalpies in Eq. 2.
The estimated dihedral angle lies between 70° and 130° displaying a decreasing trend with pressure for the 30.4° tilt configuration (Fig. 5). On the other hand, θ takes larger values (130 to 150°) remaining roughly insensitive to pressure for the 49.6° configuration. If θ is lower than the critical value of 60°, even small amounts of melt can establish an interconnected network along the grain edge and corner and percolate through silicate matrix, that is, complete wetting may occur. Our prediction of systematically higher θ than the critical angle implies that the Mg2SiO4 melt does not wet the solid forsterite and rather it may remain locally trapped as patches at the grain corner or on the grain boundary. Only when melt is present in sufficiently large amount, melt connectivity and percolation become effective for θ > 60°. The required minimum volume fraction of melt for percolation to occur can be estimated as \({f}_{m}\sim 0.009\sqrt{\theta -60}\) (Dong et al. 2019). For the calculated dihedral angles of 70 to 150°, fm varies from ~ 2 to 8%. It is thus possible that partial melts present at a depth in any smaller amount than 2 volume percent form isolated pockets and hence do not significantly influence the physical properties, including elasticity and electrical conductivity of the host rock. This means that these melts in the interior may undergo undetected by geophysical observations of low seismic velocity and high electrical conductivity regions of the mantle. Alternatively, velocity or conductivity anomalies detected at depth may correspond to the regions comprising of partial melts in large amounts (a few volume percent) or should have some other origins.

Estimated dihedral angle of the forsterite–melt system as a function of pressure at 1500 K for the 30.4° and 49.6° tilt configurations (filled circles and diamonds, respectively). The horizontal line marks the critical dihedral angle of 60°. The experimental data for forsterite or olivine grains interfaced with different fluids and silicate melts at 1073 to 1473 K are shown by different symbols: triangle (Watson and Brenan 1987), plus (Mibe et al. 1998a), cross (Mibe et al. 1998b), square (Maumous et al. 2004), and asterisk (Yoshino et al. 2007)
Our calculated results show that the dihedral angle between forsterite and melt decreases with pressure is consistent with the experimental findings by (e.g., Mibe et al. 1998a; Yoshino et al. 2007). However, the absolute values of dihedral angles calculated in this study are substantially higher than the values reported by these experimental studies. A likely cause for this disagreement is the difference in the composition of the melt. In the present study, the “melt” is amorphous olivine (Mg2SiO4), whereas in Mibe et al. (1998a) it is basaltic to peridotitic and in Yoshino et al. (2007) it is nearly aqueous fluid at low pressure that changes to more silicate-rich melt at higher pressures. And both experimental studies reported that the dihedral angle decreases with pressure that corresponds to a change in the composition of the melt. However, because we do show the decrease of the dihedral angle with pressure for the same composition, we suggest that the experimentally observed pressure effects on the dihedral angle should, at least, partly be caused by the intrinsic effect, namely, the pressure variation of the solid–liquid/solid–solid interfacial energy ratio for a fixed composition.
In the simulated bi-crystal system, the structural distortion in the vicinity of the boundary plane is the consequence of lattice mismatch between the adjacent grains because of their different orientations. The interfacial atoms experience not necessarily equal but opposing potential field and these atoms undergo relaxation accordingly. The interfacial relaxation appears to be complete in our simulations. The value of \({\gamma }_{\text{SS}}\) obtained by subtracting the bulk crystalline contribution is reliably constrained because the origin of the excess energy is due to the distortion only in the vicinity of the boundary plane. This perhaps is manifested by the predicted smooth pressure variations of \({\gamma }_{\text{SS}}\) at 0 and 1500 K unlike considerably scattered values of \({\gamma }_{\text{SL}}\) (Fig. 4). The evaluation of interfacial energetics of the two-phase system is rather complicated because of the competing effects of the crystal half and amorphous half on the interfacial region and the kinetic barrier to the relaxation. While the effect of the crystal half is to bring crystalline order on the interfacial atoms, the effect of the amorphous half is to resist that ordering. These effects together with the kinetic barrier may hinder sufficient relaxation of the interfacial region. In particular, the liquid side near the interface plane seems to undergo minimal local relaxation in a way that is different than that in the bulk liquid. This means that the entire half of the system remains fully disordered (the liquid part). The solid side near the interfacial plane is highly distorted and seems to imply a disorder-intruded region. Also, the bulk of the crystal part seems to be notably distorted compared to a perfect forsterite crystal as discussed in Sect. 3.1. These features together may result in higher internal energy of the simulated solid–liquid system, thereby implying possible overestimation of \({\gamma }_{\text{SL}}\). An accurate estimation of the amorphous and crystalline contributions in crystal–melt system likely narrows the gap between the calculated and experimental values of the dihedral angle. However, it is not trivial to accurately constrain these contributions with the present methodology.
Conclusions
We report a comparative study of the structural and energetics behavior of crystal–melt (solid–liquid) and grain boundary (solid–solid) interfaces in Mg2SiO4 forsterite at upper mantle pressure–temperature conditions by performing large-scale first-principles molecular dynamics simulations. The calculated Si/Mg–O coordination environments and bond distributions in the vicinity of the interface plane of the two-phase system correspond to much more distorted structure (being almost disordered like liquid configuration) than the intergranular interfacial region. The formation enthalpy (\({\gamma }_{\text{SS}}\)) of two (0l1)/[100] grain boundaries (corresponding to tilt angles of 30.4° and 49.6°) increases almost linearly with pressure taking values between 0.8 to 1.4 J/m2 between 0 to 16 GPa at 1500 K. The calculated results are generally consistent with the experimental data. The solid–liquid interfacial enthalpy (\({\gamma }_{\text{SL}}\)) takes comparable values of 0.9 to 1.5 J/m2 with no clear pressure effect. These interfacial enthalpies allow us to understand how the grain boundary and crystal–melt interfaces compete in the forsterite–melt system.
We find that the estimated values of 67° to 146° for the liquid–solid–solid dihedral angle are larger than experimentally inferred values for most silicate systems.
Details of the discrepancy between the computational and experimental results may be attributed partly to the current limitations of the first-principles approach and partly to the other factors, such as melt composition. When studying solid–liquid interfaces a great deal of effort should be put forward to identify and account for contributions to the interfacial structure and energetics. For instance, the evaluation of \({\gamma }_{\text{SL}}\) also requires the amorphous energetics that should be very similar to that of the liquid part of the simulated two-phase system. Given that relaxation of structural motifs in the liquid depends on the short and medium range scales and proceeds slowly, it may not be feasible to obtain and subtract exact energetics for the amorphous part. Moreover, the crystal part of the simulated two-phase system seems to be excessively distorted, more so closer to the interfacial plane. Examining these issues further is beyond the scope of this work mainly because of high computing cost associated with needed much larger and longer simulations. However, it is our understanding that a careful consideration in the future can lead to reliable results for crystal–melt interfacial energies and this first-principles study represents a positive step in this endeavor.
Data availability
The data that supports the findings of this study are available within its supplementary material and additionally available from the corresponding author upon reasonable request.
References
Adjaoud O, Marquardt K, Jahn S (2012) Atomic structures and energies of grain boundaries in Mg2SiO4 forsterite from atomistic modeling. Phys Chem Min 39:749–760
Belmonte D, Ottonello G, Zuccolini MV, Attene M (2017) The system MgO-Al2O3-SiO2 under pressure: a computational study of melting relations and phase diagrams. Chem Geol 46:54–64
Benet J, MacDowell LG, Sanz E (2015) Interfacial free energy of the NaCl crystal-melt interface from capillary wave fluctuations. J Chem Phys 142:134706
Cmíral M, Fitz Gerald JD, Faul UH, Green DH (1998) A close look at dihedral angles and melt geometry in olivine basalt aggregates: a TEM study. Contrib Mineral Petrol 130:336–345
Cooper R, Kohlstedt D (1982) Interfacial energies in the olivine–basalt system. In: Akimoto S, Manghnani M (eds) High pressure research in geophysics. Adv Earth Planet Sci, Center for Academic Publications 12: 217–228
de Koker NP, Stixrude L, Karki BB (2008) Thermodynamics, structure, dynamics, and freezing of Mg2SiO4 liquid at high pressure. Geochim Cosmochim Acta 72:1427–1441
de Koker NP, Karki BB, Stixrude L (2013) Thermodynamics of the MgO–SiO2 liquid system in Earth’s lowermost mantle from first principles. Earth Planet Sci Lett 361:58–63
de Leeuw NH, Parker SC, Catlow RA, Price GD (2000) Proton containing defects at forsterite 010 tilt grain boundaries and stepped surfaces. Am Mineral 85:1143–1154
Dong J, Li J, Zhu F (2019) Wetting behavior of iron–carbon melt in silicates at mid-mantle pressures with implications for Earth’s deep carbon cycle. Front Earth Sci 7:268
Duyster J, Stöckhert B (2001) Grain boundary energies in olivine derived from natural microstructures. Contrib Mineral Petrol 140:567–576
Ghosh DB, Karki BB (2014) First principles simulations of the stability and structure of grain boundaries in Mg2SiO4 forsterite. Phys Chem Miner 41:163–171
Ghosh DB, Karki BB, Wang J (2022) Hydration-driven stabilization and volume collapse of grain boundaries in Mg2SiO4 forsterite predicted by first-principles simulations. Am Mineral 107:744–753
Gleason GC, Bruce V, Green HW (1999) Experimental investigation of melt topology in partially molten quartzo-feldspathic aggregates under hydrostatic and nonhydrostatic stress. J Metamorp Geol 17:705–722
Hier-Majumder S, Richard Y, Bercovici D (2006) Role of grain boundaries in magma migration and storage. Earth Planet Sci Lett 248:735–749
Hiraga T, Anderson IM, Kohlstedt DL (2004) Grain boundaries as reservoirs of incompatible elements in the Earth’s mantle. Nature 427:699–703
Holness MB (2006) Melt-solid dihedral angles of common minerals in natural rocks. J Petrol 47:791–800
Holness MB (2010) Decoding dihedral angles in melt-bearing and solidified rocks. J Virtual Explorer, Electronic Ed., ISSN 1441–8142, volume 35, paper 2 In: (Eds.) MA Forster and John D Fitz Gerald, The Science of Microstructure—Part I
Jurewicz SR, Watson EB (1985) The distribution of partial melt in a granitic system: the application of liquid phase sintering theory. Geochim Cosmochim Acta 49:1109–1121
Karki BB, Ghosh DB, Verma AK (2015) First-principles prediction of pressure-enhanced defect segregation and migration at MgO grain boundaries. Am Mineral 100:1053–1058
Kato T, Kumazawa M (1985) Effect of high pressure on the melting relation in the system Mg2SiO4-MgSiO3 Part I. Eutectic relation up to 7 GPa. J Phys Earth 33:513–524
Kresse G, Furthmüller J (1996) Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys Rev B 54:11169–11186
Kresse G, Joubert D (1999) From ultrasoft pseudopotentials to the projector augmented-wave method. Phys Rev B 59:1758–1775
Laird BB, Davidchack RL (2005) Direct calculation of the crystal-melt interfacial free energy via molecular dynamics computer simulation. J Phys Chem B 109:17802–17812
Marquardt K, Faul UH (2018) The structure and composition of olivine grain boundaries: 40 years of studies, status, and current developments. Phys Chem Miner 45:139172
Maumus J, Laporte D, Schiano P (2004) Dihedral angle measurements and infiltration property of SiO2-rich melts in mantle peridotite assemblages. Contrib Mineral Petrol 148:1–12
Mibe K, Fujii T, Yasuda A (1998a) Connectivity of aqueous fluid in the Earth’s upper mantle. Geophys Res Lett 25:1233–1236
Mibe K, Fujii T, Yasuda A (1998b) Control of the location of the volcanic front in island arcs by aqueous fluid connectivity in the mantle wedge. Nature 25:1233–1236
Perdew JP, Burke K, Ernzerhof M (1996) Generalized gradient approximation made simple. Phys Rev Lett 77:3865–3868
Pommier A, Kohlstedt DL, Hansen LN, Mackwell S, Tasaka M, Heidelbach F, Leinenweber K (2018) Transport properties of olivine grain boundaries from electrical conductivity experiments. Contrib Mineral Petrol 173:41
Presnall DC, Walter MJ (1993) Melting of forsterite, Mg2SiO4, from 9.7 to 16.5 GPa. J Geophys Res Solid Earth 98:19777–19783
Presnall DC, Wen YH, Milhooland CS, Walter MJ (1998) Liquidus phase relations in the system MgO–MgSiO at pressures up to 25 GPa—constraints on crystallization of a molten Hadean mantle. Phys Earth Planet Inter 107:83–95
Smith CS (1948) Grains, phases, and interfaces: an interpretation of microstructure. Trans Metall Soc AIME 175:15–51
Terasaki H, Frost DJ, Rubie DC, Langenhorst F (2005) The effect of oxygen and sulphur on the dihedral angle between Fe–O–S melt and silicate minerals at high pressure: implications for Martian core formation. Earth Planet Sci Lett 232:379–392
Toramaru A, Fujii N (1986) Connectivity of melt phase in a partially molten peridotite. J Geophys Res 91:9239–9252
Waff HS, Bulau JR (1979) Equilibrium fluid distribution in an ultramafic partial melt under hydrostatic conditions. J Geophys Res 84:6109–6114
Walker D, Jurewicz S, Watson EM (1988) Adcumulus dunite growth in a laboratory thermal gradient. Contrib Mineral Petrol 99:306–319
Watson EM, Brenan JM (1987) Fluids in the lithosphere, 1. Experimentally-determined wetting characteristics of CO2–H2O fluids and their implications for fluid transport, host–rock physical properties, and fluid inclusion formation. Earth Planet Sci Lett 85:497–515
Yoshino T, Nishihara Y, Karato S-I (2007) Complete wetting of olivine grain boundaries by a hydrous melt near the mantle transition zone. Earth Planet Sci Lett 256:466–472
Zhimulev EI, Chepurov AI, Sonin VM, Litasov KD, Chepurov AA (2018) Experimental modeling of percolation of molten iron through polycrystalline olivine matrix at 2.0–5.5 GPa and 1600 °C. High Press Res 38:153–164
Acknowledgements
This research was supported by NSF (EAR 1463807). High computing resources were provided by Louisiana State University.
Author information
Authors and Affiliations
Contributions
BBK and DB designed research and performed simulations. All authors contributed to the analysis and discussion of the results and to the preparation of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Karki, B.B., Ghosh, D.B., Wang, J. et al. Crystal–melt interfaces in Mg2SiO4 at high pressure: structural and energetics insights from first-principles simulations. Phys Chem Minerals 50, 31 (2023). https://doi.org/10.1007/s00269-023-01256-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00269-023-01256-3