
Abstract
Computer simulation and modeling have proven to be powerful tools in the fields of engineering and polymer science. These computational methods not only enable us to verify experimentally observed behaviors, but also provide answers to unsolved phenomena. This review addresses the current status and trends of computational and theoretical studies in polymer blends. We briefly discuss the fundamental aspects of polymer blends, including experimental observations, theories, and a variety of molecular simulations and models for mixtures of two or more polymeric materials. In particular, this study deals with the description of coarse-grained techniques that can offer perspectives into the collective behavior and properties of complicated systems. Additionally, a detailed analysis of their structural, rheological, and mechanical properties via computation is also examined. Lastly, we summarize important findings and highlight points to be carefully considered in modeling polymer blends system accompanied by an outlook on the extension of current studies to complicated systems of many blending types.
Similar content being viewed by others


1 Introduction
Computer simulation is of great importance in science and engineering fields along with experiments and theories. This computational approach enables us to predict the structure, dynamics, and other material properties of complex macromolecules. With significant advances in computation, there have been considerable efforts in polymer modeling and simulation from the atomistic to the macroscopic level [1, 2]. The fully atomistic or united atom model (considering specific units as one site) [3] is used to describe detailed atomistic information of nanomaterials [4, 5]; however, it is unfeasible for large-scale applications, due to a significant amount of computing time cost. Mapping atomistic into coarse-grained (CG) representation (as illustrated in Fig. 1) can, therefore, extend to complicated macromolecular systems by simplifying the physical system through a reduction in degrees of freedom.

Schemes illustrating mapping atomistic representation into CG beads
With the development of suitable mapping schemes, they can still capture the fundamental physical characteristics of phenomena observed in high-resolution models or experimental data [6,7,8]. This CG simulation offers an attractive opportunity to describe complicated phenomena, such as phase separation [9], crystalline behavior [10], biomolecules [11, 12], self-assembly [13, 14], etc. Yet, there have been reports that inappropriate CG parameterization and methods may result in artifacts (e.g., thermodynamic inconsistency, fast dynamics, etc.) [15, 16], leading to an invalid thermodynamic expression with inexact preservation of experimental observables. Accordingly, it is a continual challenge to adopt or develop effective CG models or techniques for each system to minimize inaccuracies induced by lost atomistic details.
Polymer, a connection of repetitive monomers, has been well known to have its own internal microstructure, showing a hierarchy of different lengths and corresponding time scales [17, 18]. The time scales of the polymer span a wide range from femtoseconds to even seconds (also for length scales ranging from nanometers to millimeters or larger); thus, this cannot be described by just a single model or simulation. In this regard, multiscale molecular simulation, bridging atomistic and CG force fields, would efficiently help to directly demonstrate macroscopic properties and behaviors from underlying molecular phenomena in polymeric materials [19,20,21,22]. Particularly, this computational technique would be beneficial to understand the properties and morphologies of polymeric materials blended with multiple components. There have been many efforts to study mixtures of polymeric materials [23,24,25,26,27], which often exhibit enhanced properties compared to a single polymer. Such studies are necessary for developing the polymer industry in that mixtures can be more economical and tailor product properties. Most polymer pairs, however, are immiscible, and thus tend to show phase separation behavior, leading to deteriorated properties [28]. These thermodynamic behaviors of polymer blends significantly affect their morphology and rheological features; thus, it is crucial to identify the characteristics of interfacial properties and structures between each blend component to improve their miscibility.
The present paper is a review of the current status and trends in the field of polymer blends via computer simulation. This includes computational methodologies at various time and length scales including polymer blend models and theories. We organize the review as follows. Section 2 briefly overviews the basics of polymer blends covering experimental observations, theories, and models. Also, Sect. 3 introduces widely well-known computational methodologies used in the field of polymeric materials, from molecular scales (molecular dynamics and Monte Carlo) to macromolecular scales (coarse-graining models, dissipative particle dynamics, etc.). In Sect. 4, we discuss the structural, rheological, and mechanical properties of polymer blends, with a detailed analysis of current published experimental and computational researches. This paper also deals with how we can properly construct a polymer blend model and compute its properties based on computer simulation. Finally, we conclude the review by stating the current challenges in the modeling and simulation of polymer blend system and future direction.
2 Background: polymer blend theory
2.1 Polymer miscibility
Polymer blends can exhibit either homogenous or heterogeneous. This polymer miscibility is known to be governed by the Gibbs free energy of mixing \(\Delta G\) as defined by
where \(\Delta H\) and \(\Delta S\) are the enthalpy and entropy of mixing at a given temperature T, respectively. In Eq. (1), when the value of \(\Delta G\) is negative, then components in a mixture become miscible. Since the contribution of entropy of mixing becomes negligibly small for higher molecular weight due to the restricted number of possible configurations, \(\Delta G\) is primarily influenced by \(\Delta H\). Flory and Huggins [29,30,31] introduced the formula for the entropy and enthalpy of mixing based on the mean-field lattice model (Fig. 2), deriving an estimate of the miscibility of polymers.

Schematic drawing of the lattice chain model representing a mixture of polymers A (blue) and B (red)
This model describes a macromolecule as a random walk on a lattice consisting of N sites of equal volume. Each bead of the polymer occupies one lattice, and a bond connecting adjacent beads of the molecule represents a link of the lattice. For a binary mixture of polymer chains A and B, each consisting of NA and NB segments per chain, respectively, the researchers obtained the following expression for the change in the free energy of mixing \(\Delta F\) at a given temperature T:
Here, \(k_{B}\) is Boltzmann constant and \(\phi_{i}\) is the volume fraction of chain i, respectively, while \(\chi\) is a dimensionless quantity, so called the Flory–Huggins interaction parameter, expressed by:
where z represents the coordination number of species and εij represents the interaction energy between the species i and j, respectively. The initial two terms in Eq. (2) arise from the entropy of mixing, while the last term is based on the enthalpy of mixing. The \(\Delta F\) depends on the value of \(\chi\) parameter as well as on the degrees of polymerization of the species [31,32,33]. The \(\chi\) parameter in Eq. (3) describes the phase behavior (mixed or unmixed) of polymer–polymer or polymer–solvent systems. It represents the energetic contribution to the free energy of mixing in a polymer mixture, in other words, a parameter of the differences in the strength of interaction energies between components in a mixture. A positive value indicates favorable interaction, while a negative value denotes unfavorable interaction, resulting in phase separation or immiscibility. The Flory–Huggins parameter is related to the solubility parameter \(\delta\) [34]. The \(\delta\) is an important property that defines the ability to be dissolved, determined by the square root of the cohesive energy density CED as below.
Here, \(\Delta H_{vap}\) denotes the heat of vaporization and \(R\) denotes the gas constant, respectively, while \(V_{m}\) is the molar volume of species. According to the Hansen solubility parameter [35], CED is generated by molecular forces (e.g., dispersion force, dipole–dipole interaction, or hydrogen bond interaction), thus this often determines the degree of solubility in a mixture. The relationship between the \(\chi\) parameter and the solubility parameter is defined by
This implies that the difference in \(\delta\) of species A and B in a mixture can connote the polymer miscibility. However, the value of \(\chi\) parameter computed from the Hildebrand solubility parameter via Eq. (5) is often invalid for mixtures containing polar and hydrogen bond interaction. Instead, the Hansen solubility parameter has proven to be useful for estimating the Flory–Huggins parameter. This is expressed in terms of the three solubility parameters from dispersion forces (d), polar forces (p), and hydrogen bond interactions (h):
This is based on the idea that a molecule can be dissolved by overcoming surrounding molecular interactions. Tables 1 and 2 present the values of two solubility parameters and \(\chi\) parameter for various polymeric materials [36,37,38,39,40,41,42,43].
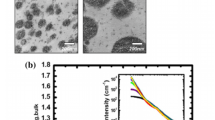
While these findings offer insightful information of miscibility, these methodologies are often inadequate for certain polymers and solvents [48, 49]. This can be attributed to intrinsic interactions such as hydrogen bonding or other complicated interactions. Furthermore, \(\chi\) parameter also shows dependence on the concentration of components as shown in Fig. 3a [50,51,52].

Behaviors of the Flory–Huggins interaction parameters with a composition and b temperature for various polymer blend: polystyrene/poly(p-methylstyrene (dPS/PpMS), PS/dPS, deuterated poly(methylbutylene)/poly(ethylbutylene) (dPMB/PEB), polypropylene/deuterated head-to-head polypropylene (PP/dhhPP), PS/polybutadiene (PB), dPS/poly(tetramethyl bisphenol A polycarbonate (TMPC), and dPS/poly(vinyl methyl ether) (PVME). Reproduced from [51]
2.2 Phase behavior of polymer blend
Empirically, it has been well established that \(\chi\) is a function of T (Fig. 3b), as shown below [31, 52]:
The temperature-independent term A contributes to the entropic part of \(\chi\), while the second term represents its enthalpic part. This observation aligns well with polymer blends [50, 53,54,55]. Generally, polymer blend systems exhibit two critical solubility temperatures: an upper critical solution temperature (UCST), which is the critical temperature for phase separation occurring at low temperatures, and a lower critical solution temperature (LCST) where phase separation occurs at higher temperatures. These two solubility boundaries define a region of miscibility gap where phase separation takes place. For miscible polymer blends, it is common for them to undergo segregation at elevated temperatures (LCST behavior), whereas UCST behavior is relatively less observed [56, 57]. This can be understood through Eq. (1). The majority of solutions have a positive value of \(\Delta S\), attributed to increased configurational entropy. This leads to a critical temperature (UCST) at which \(\Delta G\) becomes zero. However, since the value of \(\Delta H\) for most polymers is typically negative (indicating exothermic mixing), they tend to exhibit LCST behavior rather than UCST. Figure 4 presents a schematic representation of the phase diagram for polymer blends with LCST and UCST.

Schematic phase diagram of LCST and UCST blends. Binodal curve is depicted as solid line and spinodal curve as dashed line
However, it should be noted that relying solely on the negative value of the \(\Delta G\) is insufficient to determine the stability of a mixture against phase separation with varying compositions. This merely indicates that the mixture will not segregate into two distinct phases consisting of pure components. Phase separation can still occur even when \(\Delta G\) is negative; thus, it is imperative to take into account the second derivative of the free energy \(\left( {\frac{{\partial^{2} \Delta G}}{{\partial \Delta \phi^{2} }}} \right)_{P,T}\). The binodal curve (known as the coexistence curve) refers to the phenomenon of distinct nucleation and subsequent growth in small spherical domains of the second phase, which gradually increase in size over a period of time. When \(\left( {\frac{{\partial^{2} \Delta G}}{{\partial \Delta \phi^{2} }}} \right)_{P,T}\) becomes negative within the binodal boundary, it enters the spinodal region. Notably, the binodal and spinodal points coincide at the critical temperature, as depicted in Fig. 4. The regions between the binodal and spinodal points can be characterized as metastable regions, wherein minor fluctuations fail to induce phase separations.
Figure 5 shows the nonlinear behavior of \(\chi\) parameter for many polymeric materials at various temperatures [58,59,60]. In cases where nonlinearity is observed in Eq. (7), \(\chi\) can be fitted to a quadratic function of 1/T as following Eq. (8) [61,62,63].

a Flory–Huggins interaction parameter as a function of 1/T, and b the reduced value \(\chi - A - B/T - C/T^{2}\) as a function of p/T for deuterated poly(ethylbutylene)/poly(methylbutylene) (PEB/DPMB) done by Lefebvre et al. [59] The symbols represent the volume fractions of dPMB equal to 0.414 (pink and purple triangles), 0.161 (yellow and mustard diamonds), and 0.099 (orange and maroon circles) from the one-phase and equal to 0.161 (green and forest squares) and 0.099 (blue and navy circles) from the metastable at p = 0.01 and 0.86 kbar. Reproduced from [58]
There are various methods to estimate the value of \(\chi\) parameter, including techniques such as small angle X-ray and neutron scattering [59], viscometry [64], differential scanning calorimetry (DSC) [53], or gas chromatography [65] (we will discuss computational methods for calculating \(\chi\) parameter later). Each method for estimating \(\chi\) parameter has its own advantages and limitations; thus, the selection of an appropriate method for measurement should be based on the specific characteristics of the system and the objectives of the research. Moreover, combining multiple experimental approaches can often yield accurate and comprehensive results. As such, the utilization of those approaches can achieve a deeper understanding of the phase behaviors of polymer blends, leading to more robust interpretations of the experimental data.
3 Background: computer simulation of the polymer
Since their pioneering efforts in 1953 by Metropolis [66] with Monte Carlo (MC) simulations and in 1957 by Alder and Wainwright [67] with molecular dynamics (MD) simulations, computer simulation methods, and algorithms have made significant progress. MD and MC methods, based on Newtonian dynamics and statistical mechanics, facilitate the direct measurement of physical properties and phenomena at the atomistic and molecular levels. Consequently, computer simulations serve as an important tool that supplements existing experimental findings and even offers a means to explore properties that are difficult to access through experimentation alone. Diverse computational techniques corresponding various time and length scales are illustrated in Fig. 6.

Time- and length-scale representation of various computational techniques
In general, atomistic methods include MD and MC simulations, while mesoscale methods include coarse-grained (CG) simulation [12, 68, 69], dissipative particle dynamics (DPD) [70, 71], or Brownian dynamics (BD) [72]. Continuum methods, including the finite element method (FEM) [73], are governed by appropriate constitutive equations, such as the Navier–Stokes equation [74]. These methods are based on the assumption that the properties of the system can be represented by continuous fields, such as temperature, pressure, and velocity. In this section, we provide a concise overview of simulation techniques of polymeric materials, covering integration schemes, thermostats, barostats, and force fields. The discussion consists of two parts: (i) molecular scale modeling (with an emphasis on MD simulation) and (ii) macromolecular scale modeling.
3.1 Molecular scale modeling
Polymer exhibits a diverse range of phenomena spanning multiple length and time scales, facing challenges in the investigation of their intrinsic characteristics. In general, it is observed ranging from the atomic scale of monomers (angstroms) to the larger scale of polymer radius of gyration (nanometers), and time scales encompassing femtoseconds up to seconds, minutes, or even years [52]. In this regard, atomistically detailed computer-aided simulations could be a useful tool for studying the molecular mechanisms of polymers and moreover help to provide insights into underlying characteristics by bridging the gap between the atomic scale and meso/macro-phenomena. One of the most widely used simulations, MD simulation calculates the time-dependent variables of a system of particles by integrating their equations of motion as shown in Eq. (9).
Here, \(m_{i}\) is atomic mass, \({\mathbf{a}}_{i}\) is its acceleration, and \({\mathbf{F}}_{i}\) is the force acting upon particle i, in a system consisting of N particles, respectively. There are various algorithms for integrating the equations of motion: Verlet [75], Velocity Verlet [76], Leap-frog [77], Beeman’s algorithm [78], etc. The widely utilized algorithm, Velocity Verlet (as an extension of the Verlet algorithm) is a numerical algorithm in which both the position and the velocity are updated at each time step. The basic equations are as follows:
where \({\mathbf{r}}\) is the position vector, \({\mathbf{v}}\) is the velocity vector, and \({\mathbf{F}}\) is the force vector with time t, respectively. This algorithm makes it possible to achieve energy conservation, thereby reinforcing the accuracy in simulation and proving its suitability for capturing the intricate dynamics of molecules. The selection of an appropriate integration scheme in MD simulation requires a thorough consideration of the characteristics of the target system to obtain reliable results in simulation. Additionally, temperature and pressure controls are achieved by introducing additional dynamic variables into the original system, effectively acting as a thermostat (e.g., Velocity scaling [79], Nosé-Hoover [80, 81], or Langevin [82, 83]) or barostat (e.g., Berendson [84], Anderson [85]). These supplementary variables play a crucial role in regulating and maintaining the desired temperature or pressure conditions throughout the simulation.
Once a suitable molecular scale model has been identified, we should find an appropriate force-field for the polymer system from established libraries. Polymer exhibits both bonded and non-bonded interactions. Bonded interactions refer to chemical bonding including bond stretching, bond angle, and dihedral angle interactions. Non-bonded interactions, on the other hand, which include van der Waals forces, hydrogen bond interaction, and electrostatic interaction, arise from intermolecular forces between chains. Specifically, these interactions occur among loosely interacting groups of atoms. Each force-field comprises a set of parameters and functions, consisting of multiple terms with distinct characteristics, as represented in the formula for the total interaction potential \(U\) of particle i in Eq. (11).
where the first three terms represent bond, angle, and dihedral interaction energy and the other two terms are van der Waals and electrostatic interaction energy, respectively. \({\mathbf{R}}_{i}\) is the position vector of atoms in a polymeric system. There are many functional formulas for each interaction, for instance, COMPASS [86], CHARMM [87], AMBER [88], etc.; however, the details are not covered in this article.
In particular, nonequilibrium molecular dynamics (NEMD) techniques have been developed as an extension of traditional MD, providing a means to investigate significant physical properties within a system subjected to an external field, such as shear or extensional flow. Therefore, it enables us to enhance our understanding of the intricate phenomena of materials departing from equilibrium. NEMD requires thoroughly considered equations of motion along with the appropriate boundary conditions and thermostat settings for each flow field in the system. Under the assumption of applying the proper methodologies, NEMD has proven to be an effective tool for predicting transport properties such as shear viscosity, diffusivity, and thermal conductivity. The first requirement for incorporating a field into the equations of motion of the system is that it should be consistent with the periodic boundary conditions (PBCs) to maintain the homogeneity of the system. PBCs effectively mitigate surface effects that can occur in a fixed simulation box, thereby reducing the computational cost of simulations. Specifically, the simulation box can become deformed imposed by the flow field, leading to numerical instability in the simulation [89]. One well-known approach is the Lees–Edwards boundary conditions [90] which allows to investigate the bulk properties of molecules undergoing shear flow. Such a scheme involves duplicating the simulation cell through periodic images, thereby accounting for the translation of molecules, and accurately representing the boundary effects under actual flow. The Kraynik–Reinelt [91, 92] boundary conditions control the unit lattice vectors under the planar elongational flow, facilitating the remapping of the simulation cell from the extended to undeformed state.
The first NEMD algorithm, so-called DOLLS [93], was suitable in the limit of linear strain rates, but exhibited remarkable errors in properties at high flow fields [94]. Therefore, Evans and Morriss [95] and Ladd [96] proposed the SLLOD algorithm for planar Couette flow which has been shown to be exact for all values of the flow field. Also, Hoover et al. [97] explored the validity of both the DOLLS and SLLOD algorithms in conjunction with boundary conditions for simple shear. According to Baig et al. [98], they developed p-SLLOD algorithm, which is compatible with Lagrangian and Hamiltonian mechanics [99]. It contains the following equations of motion.
where \({\mathbf{q}}_{i}\) and \({\mathbf{p}}_{i}\) are the position vector and the peculiar momentum vector (except external flow field), whereas \(m_{i}\) and \({\mathbf{F}}_{i}\) are the mass and the force vector of atom i, respectively. \({\mathbf{u}}\) is the streaming velocity. They demonstrated its capability to model the system under both shear and extensional flow, enabling the direct calculation of a variety of structural, thermodynamic, and rheological properties.
As previously mentioned, the use of inappropriate algorithms or thermostatting techniques can lead to the artifacts, such as unstable values of properties, energy non-conservation, or convergence problems during NEMD simulation. Delhommelle et al. [100] addressed the occurrence of unphysically observable phenomena under an assumed flow profile. The work stressed the importance of employing a configurational thermostat (without assumption of flow field in NEMD) to realistically represent the system under flow conditions. In line with the findings by Delhommelle et al. Baranyai [101] also demonstrated anisotropy between configurational and kinetic temperatures. Li et al. [102] stressed that it should be employed to different formulations for thermal transport depending on the thermostat method through a comparison between the Nosé–Hoover thermostat with the Langevin approach. Additionally, Ke et al. [103] conducted a comprehensive investigation into the widely used thermostats and barostats in terms of accuracy in physical properties and fluctuations, thus emphasizing on the importance of considering effective thermostats/barostats in contrast with equilibrium MD. To prevent issues arising from the incorrect construction of simulation methodologies, careful attention should be paid to the selection of the suitable thermostats and algorithms based on relevant literatures. Furthermore, it is needed to meticulously organize NEMD simulation strategies depending on the specific characteristics of the systems for the desired physical properties. As pointed by Anstine et al. [104], they estimated the mechanical properties of glassy polymers, concluding that physical properties of polymers can be significantly influenced by modeling techniques, such as force-field and polymer builder. Hence, one has to be very cautious in applying thermostats/barostats in conjunction with other algorithms in NEMD simulation to accurately estimate values of physical properties.
The MC technique, also known as the Metropolis method [66], involves generating a sample of the population using random numbers. The key steps in MC simulation can be summarized as follows: (i) Formulating the physics problem of interest into a probabilistic model. (ii) Numerically sampling the probability model to obtain representative configurations. (iii) Analyzing the collected data using statistical models. The fundamental principle of MC involves estimating the average characteristics of system based on the probability distribution, associated with the ensembles in statistical mechanics. While MD simulations track the evolution of a system over time, MC simulations enable the estimation of ensemble-averaged properties through statistical sampling. By employing random sampling, MC simulations efficiently explore the phase space and provide valuable information in equilibrium thermodynamics and statistical mechanics.
3.2 Macromolecular scale modeling
CG methods are based on hierarchies of overlapping length or time scales, where the majority of degrees of freedom are reduced by grouping or averaging atoms into larger entities. These models aim to capture the essential features and collective behavior of the system while reducing computational costs. In CG, multiple atoms are typically combined into a single interaction site, referred to as a CG particle or bead. These beads represent a collection of atoms with similar properties and behavior. The interactions between beads are described by effective potentials which can unveil the thermodynamic and structural characteristics of the system at the CG level. Similar to atomistic models, bonded potentials within a CG bead can be depicted by several empirical models, such as finite extensible nonlinear elastic (FENE) bond potential [105]. The FENE potential follows the expression:
where \(R_{0}\) is the maximum bond extension length, \(k\) is the spring constant, and \(R_{ij}\) is the bond vector between particle i and j, respectively.
Most commonly used CG models in polymer systems are Martini [106, 107], united-atom [3], Kremer–Grest (KG) [105, 108], etc. The Martini model is mainly employed for biosystems such as proteins and lipids. This model typically makes a group of four heavy atoms into one CG bead, allowing for the investigation of large-scale conformational changes, self-assembly processes, and membrane behaviors [13, 109, 110]. Furthermore, this model with its high computational efficiency, is suitable for calculating interactions within polymer blends, even those involving large molecules [111,112,113]. The united-atom model simplifies the representation of molecules by merging hydrogen atoms with their adjacent heavy atoms. In other words, hydrogen atoms are treated as parts of the heavy atoms they are bonded to, effectively reducing the total number of particles in the system. This model has been extensively employed in simulating polymers, enabling investigation into phase transition, diffusion processes, and other dynamic characteristics [114, 115]. The KG model, a bead-spring model, represents a chain as successive beads connected by springs. Each bead interacts with neighboring beads through potential energy function (e.g., Weeks–Chandler–Anderson interactions [116], FENE potential, or a bending potential introduced by Faller and Müller-Plathe [117,118,119]) including the bond, angle, and dihedral interactions. One of the key features of the KG model is its ability to simulate entanglements and topological constraints between polymer chains; thus, it can express the realistic behavior of polymers, including their conformational changes, chain relaxation, and diffusion dynamics. Consequently, the model is effective for studying various aspects of polymer blends, such as melt dynamics, phase transition, or miscibility [120, 121].
Dissipative Particle Dynamics (DPD) [70, 71] is a mesoscale simulation method that has gained significant popularity in the field of soft matter and complex fluids. In DPD, particles represent groups of atoms, and interactions are governed by conservative (c), dissipative (d), and random forces (r) between particle i and j as described by Eq. (14).
Here, the conservative force represents long-range interactions, while the dissipative force dissipates energy, thereby describing the effects of viscosity. The random force accounts for thermal fluctuations. DPD has been applied to studying self-assembly processes, micellar systems, and flow behavior, offering the details of collective behavior and dynamics of complex systems [122, 123].
Brownian Dynamics (BD) simulations [72] are stochastic simulation methods that focus on modeling diffusion processes. In contrast to MD simulations, BD operates on a much larger time scale, typically up to a few nanoseconds. It is commonly employed in investigating solvent–polymer interactions using an implicit continuum solvent description. The governing equation in BD, known as the Langevin equation [82, 83], incorporates dissipative and random force terms that account for the effects of polymer–solvent interactions, including the influence of solvent removal from the polymer. The Langevin equation can be expressed as:
where \(\gamma\) is the friction coefficient, \(p_{i}\) is the momentum, and \(\xi_{i} (t)\) is the random force with the coupling coefficient \(\sigma\) of the particle i at time t, respectively. By employing BD simulations, researchers can investigate the diffusion and mixing behavior of polymer blends [124]. The motion of each polymer chain is influenced by both conservative and stochastic forces, describing the effects of the surrounding solvent and thermal fluctuations. Based on this algorithm, it allows us to predict the macroscopic properties of polymer blends, such as viscosity, diffusion coefficients, and phase separation behavior [125].
The aforementioned CG techniques, which go beyond atomistic simulations, are valuable approaches in molecular modeling, offering perspectives into the collective behavior and properties of complicated systems. Therefore, it is desirable to perform multiscale simulations by combining atomistic and CG scale models to demonstrate phenomena occurring across various scales. This approach contributes to a deeper understanding of molecular system behavior.
4 Characteristics of the polymer blend
4.1 Morphology and miscibility
Over the years, it has been widely reported that polymers do not readily mix to form a homogeneous mixture. Accordingly, their morphologies significantly dictate the physical properties of these phase-separated polymers, such as toughness, flowability, and optical properties. Therefore, it is crucial to establish the relationship between them and to predict the morphology. Recent advancements in microscopy techniques, including scanning electron microscopy (SEM), atomic force microscopy (AFM), and transmission electron microscopy (TEM) have allowed for more detailed observations of the morphology of polymer blends.
For high-molecular-weight polymer blends, miscibility is primarily dependent on the values of \(\Delta H\) and \(\chi\) parameter, due to the low contribution of the entropy of mixing as previously mentioned in Eq. (1). There have been numerous studies aiming to quantify the \(\chi\) parameter of polymer blends. Lee et al. [126] determined the miscibility for polystyrene (PS)– polybutadiene (PBD) blends investigated by DSC. They investigated values of the Flory–Huggins interaction parameter, which ranged from 0.0040 to 0.0102, for different molecular weights and compositions of the PS–PBD blends. Roe et al. [127] also calculated the \(\chi\) parameter of PS–PBD blends under various conditions including temperature, via light scattering measurement. Additionally, the surface morphology of UCST blend [isotactic polystyrene (iPS)–poly(4-methyl styrene) (P4MS) blend] was studied by Chang and Woo [128] through AFM and DSC. The values of \(\chi\) parameter for Ips–P4MS blends are found to be in the range of 0.0163 – 0.0232 depending on the composition. All these values are greater than the values at the critical points, indicating that iPS–P4MS blends are immiscible for all compositions. Such immiscible or miscible phenomena are closely correlated with the interface of morphology between each component in a mixture. The study conducted by Li et al. [129] investigates the role of the interface in the morphologies of three different categories of blend: (i) high-density polyethylene (HDPE)/styrene–ethylene–butylene–styrene (SEBS) and HDPE/styrene–ethylene–butylene (SEB), (ii) an incompatible binary system comprised of HDPE/PS, and (iii) compatible ternary systems comprised of HDPE/PS compatibilized by SEBS and SEB, respectively. The results conclude high interfacial tension shows a strong dependency on the composition of pore size of co-continuous microstructure. According to Taguet et al. [130], the detailed interface–morphology relationship in PE–thermoplastic starch (TPS) blends is elucidated by the formation of a thin glycerol-rich layer at the PE–TPS interface, which plays a role as an interfacial modification. They added the quantity of poly(ethylene-g-maleic anhydride) (PE-g-MA) copolymer (a compatibilizer) allowing to place itself at the interface of PE–TPS and improve adhesion between phases. Therefore, they suggested a potential approach to modify interfaces in polymer blend using plasticized polymers and small molecule modifiers.
There are several efforts to supplement the miscibility parameter. Tambasco et al. [51] suggested the temperature- and concentration-independent parameter, which would be more characteristic indicator of blend system than the \(\chi\) parameter, by analyzing experimental data from neuron scattering, cloud point curves, and pressure–volume–temperature (PVT) surfaces for several blends [131,132,133,134,135,136,137,138]. Based on the Born–Green–Yvon (BGY) theory [139], the authors attempted to quantify the binary interactions of polymer blends, offering a feasible substitute for the Flory–Huggins interaction parameter.
Since these experimental methods are quite complicated, many groups have devoted to observing the morphology and interaction parameters based on semi-empirical approaches or computer simulations [140,141,142]. Estridge and Jayaraman [143] performed CG simulations of AB diblock copolymer grafted particles in an immiscible A–B blend (DBCGPs) (Fig. 7).

Representative snapshots (Top and side view) of interfacial configurations with different volume fractions of DBCGP equal to a, f 7.3 \(\times\) 10–4, b, g 3.6 \(\times\) 10–3, c, h 7.3 \(\times\) 10–3, d, i 1.0 \(\times\) 10–2, and e, j 1.4 \(\times\) 10–2. Reproduced from [143]
Polymers are represented as a bead-spring model with the Weeks − Chandler − Andersen (WCA) [116] and 6–12 Lennard–Jones (LJ) [144, 145] potentials. This study has revealed that DBCGPs are effective alternatives to AB diblock copolymers (BCPs), which are well known as compatibilizers in a blend of A and B polymers. The researchers calculated the probability of DBCGP distribution at the blend interface, desorption energy, and interfacial tension. The results show a higher desorption energy and a reduction in the interfacial tension compared to that of BCPs. Additionally, Kozuch et al. [146] performed MD simulations of bead-spring chains with different stiffness, demonstrating good agreement with experimental values by mapping real polymer chains onto their bead-spring chains.
Here, it is necessary to focus on studies that combine atomistic and mesoscale simulations to accurately compute the miscibility and morphology of polymer blends [22]. The study reported by Luo and Jiang [147] investigates the miscibility of poly(ethylene oxide) (PEO)–poly(vinyl chloride) (PVC) blends via both atomistic MD and mesoscale DPD. They determined the Flory–Huggins interaction parameter from CED and intermolecular radial distribution function, concluding that a larger degree of hydrogen bonding interaction was observed in PEO–PVC blends with a weight ratio at 70/30 and 30/70 compared to 50/50, as shown in Figs. 8 and 9. These features are supported by the morphologies of PEO–PVC blends, where the 50/50 blend exhibits a weak phase separation.


Radial distribution functions of the intermolecular carbon–carbon pairs of like- and unlike-components in PEO–PVC blends with corresponding morphologies from DPD simulation (PEO: red, PVC: green). Reproduced from [147]
In addition, Spyriouni and Vergelati [148] conducted the molecular simulations at the atomistic and mesoscopic level for the binary blend of polyamide 6 (PA6) with poly(vinyl alcohol) (PVOH), poly-(vinyl acetate) (PVAC), and partially hydrolyzed PVAC for a wide range of compositions. They calculated CED of the pure components and mixture from the atomistic simulation, and then derived the \(\chi\) parameter as a function of composition for blends. Moreover, the interaction parameter, along with structural parameters, was applied to the CG simulation, thus enabling the examination of the behaviors of high-molecular-weight blends and long-time scales that allowed the observation of phase separation. For all mixtures, the highest \(\chi\) parameters were observed for the 50/50 composition. The PVAC–PA6 mixtures had the smallest, while the highest values were observed for PVOH-PA6. This is attributed to the low hydrolysis degree, in other words, a reduction in the hydrogen bonds and an increase in the portion of extended conformations. In this way, it is evident that applying the parameters obtained through atomistic-level simulations to mesoscopic-level simulations would provide substantial information and profound considerations in the morphology of polymer blends.
4.2 Rheology
Generally, quantifying the rheology of polymer blends is challenging due to the presence of viscoelastic and nonlinear effects within multiphase systems. As mentioned earlier, their structures and interactions play a crucial role in determining their rheological properties. Specifically, the proportion of components in a blend significantly influences its elasticity, viscosity, and flow behavior [149]. Moreover, the formation of networks between different phases can profoundly impact the rheological characteristics [150]. Thus, it is evident that the rheological characteristics of polymer blends are predominantly controlled by their morphology, which is distinct from that of homopolymers. Several researches have been devoted to demonstrating the rheology of polymer blends. According to Hamad et al. [151], the rheological and mechanical properties of poly(lactic acid) (PLA)–PS blends were investigated for different ratios of each component. The results show that PLA–PS blend exhibits a shear-thinning behavior. Also, the viscosity of the blend decreases with an increasing composition of PLA, which is attributed to the complicated molecular structure (high viscosity) of PS. Huang et al. [152] conducted a study that phase behavior could be regulated by altering the blend composition and, in particular, the deformation or stress history. These findings are analogous to the research reported by Peng et al. [153], wherein blend morphologies significantly affect the rheological properties. It was elucidated by conducting MD simulations on two distinct blends: a well-mixed blend and a phase-separated blend (Fig. 10a). They clearly revealed the effect of characteristics of blend components (chain length) on the diffusion and storage modulus (Fig. 10b and c). Notably, it was observed that stiff chains had the ability to slow down the dynamics of soft chains, resulting in a higher storage modulus of homogenously mixed blends, as shown below in Fig. 10c.

a Snapshots of mixed (left) and unmixed (right) blend configurations. Red spheres represent A chains (stiffer chains using FENE, LJ, and many-body interaction), and blue spheres represent B chains (softer chains using only FENE). b Plot of the center of mass (COM) and monomer (bead) mean-square displacements (MSD) of B chains in B melts and A–B blends. c Storage modulus as a function of shear frequency for A melts (black), B melts (green), mixed A–B blend (red), and unmixed A–B blend (blue). Reproduced from [153]
Iza et al. [154] found that interfacial modification significantly affected the rheological behaviors of PS–PE immiscible blends under transient flow. More precisely, it was observed that the quantity and structure of the compatibilizer, capable of modifying the blend morphology, had a significant impact on the rheological response to a suddenly imposed shear rate. Also, Huang and Hua [155] performed mesoscale simulations of Nylon 6–elastomer (ACM) blends, revealing an impact of flow history on the blend morphology. They clearly provided detailed information on the relationship between flow-induced morphology and blend rheology by computing the stress relaxation and topological properties. Wang et al. [156] studied the storage modulus and viscosity for PP–ultrahigh molecular weight polyethylene (UMWPE) blends. By combining DPD simulations and experimental measurements, it was explored that changes in the morphology led to a decrease in the miscibility and storage modulus under extension–shear coupled flow, showing good agreement with experimental results. Furthermore, Hajizadeh et al. [157] investigated the structure and rheology of the linear–dendrimer polymer blends under planar elongational flow via nonequilibrium MD simulations, revealing the effect of blend structure on rheology. Besides, there are other studies conducted on rheological properties of Polymethyl methacrylate (PMMA) – PS [158], poly(3-hexylthiophene) (P3HT)–[6, 6]-phenyl-C61-butyric acid methyl ester (PCBM) [159], protein–water or polymer [160, 161], n-alkanes–polymers [162], or poly(vinylphenol)–poly(ethylene terephthalate) [163], PS–polyisoprene [164] blends, etc.
Simulating the deformation of polymer blends under external forces requires a multiscale approach; thus, several studies have endeavored to explore the rheology of polymer blend through multiscale simulation [152, 155, 156, 159, 165]. Deng et al. [165] investigated the properties of polymer blends with shear processing by employing a combined simulation. The model systems here corresponded to PS-b-polyacrylonitrile (PAN) block copolymer systems. They observed that the morphology became anisotropic structure with applied shear stress. This phenomenon aligned with the results of normal stress field (Fig. 11), wherein the stress was relieved by the soft phase.

Normal stress field in x–z plane of PAN block copolymer blends for shear rates equal to a 0, b 2 \(\times\) 103 s−1, c 1 \(\times\) 104 s−1, and d 2 \(\times\) 105 s.−1. Corresponding morphologies are shown on the top-left corner (the hard phase is displayed in red, and the soft matrix is colored blue). Reproduced from [165]
The rheological properties of the polymer blends are significantly involved with polymer entanglements or dynamic behaviors. O’Connor et al. [166] conducted a study on the entanglement topology and nonlinear rheology of ring-linear polymer blends with varied ring fractions as depicted in Fig. 12a. Based on the primitive path analysis [167, 168], the authors quantified the degree of ring–linear threading and linear–linear entanglement. These computations then were utilized to validate rheological properties. Interestingly, the study observed an extensional stress overshoot for different ring fractions, as shown in Fig. 12b. They demonstrated that this phenomenon was attributed to overstretching and recoil of ring polymers caused by unthreading from linear chains. This implies that simply changing composition allows the stress overshoot to be varied for nonlinear strain rates.

a Primitive path chain topologies during extensional flow for linear (red)–ring (green) blends at different volume fraction of ring polymer \(\phi_{R}\) = 30% (top) and 88% (bottom). Configurations are shown at equilibrium (left) and the overshoot strain \(\varepsilon_{H}\) = 3.0 (center) and 7.5 (right). b Stress curves (at the same \(\dot{\varepsilon }_{H}\)) under startup extensional flow for blends and pure melts. Reproduced from [166]
Young et al. [125] also investigated the influence of topological constraints on the rheology of ring–linear blends. They showed ring polymers in the blend exhibited considerable conformational fluctuation, leading to a stress overshoot.
4.3 Mechanical properties
It is generally known that combining various polymers become enables the achievement of a variety of mechanical properties that are not easily attainable with individual polymers alone. These blends can exhibit distinctive behaviors, such as enhanced tensile strength, improved impact resistance, increased flexibility, and enhanced durability; whereas, improper blending can lead to phase separation or reduced mechanical performance. Hence, it would be worthwhile to understand the relationship between the molecular factors and mechanical properties of the polymer blend. Ge et al. [169] demonstrated the mechanical weakness of the interface of immiscible polymer blends via large-scale MD simulations. According to their results, the lack of entanglements at the interface induces chains to be easily pulled out from the opposite side; thus, there is a minimum interdiffusion depth required for significant entanglement formation which enables to enhance an interfacial strength. Huang and Hua also [155] provided an in-depth understanding of blend elasticity for immiscible Nylon 6–elastomer (ACM) blends based on interfacial relaxation analysis. According to study done by Salahhoori et al. [170], using MD simulations, the blend compatibility, miscibility, and mechanical behavior of poly (vinyl fluoride) (PVF), poly (vinyl chloride) (PVC), and poly (vinyl acetate) (PVAc) with Pebax-1657 (Px) were examined. They calculated the shear modulus, bulk modulus, and Young’s modulus under different stress conditions. Researchers demonstrated that due to the chemical bonding of Px with PVAc, PVC, and PVF, miscibility was improved, and notably, the Px–PVC blend effectively formed a cross-linked network, resulting in enhanced mechanical properties. Also, Wei et al. [171] attempted to compute the mechanical properties of nature rubber-graft-rigid-polymer and rigid-polymer blend systems based on computational approaches. Their results showed that a higher strain rate gave rise to a higher stress modulus or yield stress. They also found that systems with increasing internal structure factors, such as nonbonding interaction or architecture (length and the number of graft chains), exhibited a better mechanical performance (Fig. 13).

a Stress–strain curves for various strain rates (at the interaction parameter within rigid-polymer beads \(\varepsilon_{RR}\) = 5.0) and b tensile modulus as a function of \(\varepsilon_{RR}\) for nature rubber-graft-rigid-polymer and rigid-polymer blend systems. \(\varepsilon_{PP}\) denotes the interaction parameter within polymer. Reproduced from [171]
There is another study on mechanical characteristics where PMMA and dibutyl phthalate (DBP) binary systems are simulated by MD simulation [172]. They calculated the mechanical properties, based on the Reuss mean method [173], revealing the effect of temperature and DBP ratio on Young’s modulus, bulk modulus, shear modulus, and Poisson’s ratio. The trend reflects the addition of DBP can improve the ductility of PMMA. Moreover, Zhao et al. [174] predicted the mechanical properties of A–B polymer blend film by MC simulation combined with the lattice spring model. As they applied the force perpendicular to the film surface, it was observed that the stress-field distribution closely matched the film’s morphology. Those kinds of work may help us to find useful information for designing and fabricating high-performance polymer blends.
5 Conclusion
This review summarizes the theories, simulation methodologies, and properties (morphology, rheology, and mechanical characteristics) of various polymer blend systems. Numerous studies regarding polymer blends via computational approaches from atomistic to macromolecular scale have been reported for molecular weight, molecular architecture, blending composition, or plasticizers. In general, most polymer blends used to be immiscible; thus, the degree of miscibility significantly determines the blend characteristics. Therefore, it is beneficial to employ multiscale simulations to get the molecular features of polymer blends across various scales. In most cases, computational methods easily calculate the value of \(\chi\) parameter, also correlating with the miscibility or morphology of many polymer blend systems. These show a good agreement with experimental results, and furthermore successfully associated with molecular structure predicted from the simulations. In particular, the rheological and mechanical properties of polymer blends are closely related to their morphology as well; many studies provide insight into controlling its properties. But still, there are difficulties in modeling and simulating complicated polymer blends such as multiphase blends or crystalline components; thus, significant effort has been devoted to predicting them, in terms of rheological or mechanical properties via computer simulations.
Also, one has to bear in mind, the conditions necessary for accurate molecular simulation of polymeric materials. That is, efficiently linking the atomistic and CG force fields is the key to multiscale simulation of polymer blend systems. There are many related studies mentioned; however, we carefully consider the computational methodologies for desired systems by linking atomistic and CG methods to match the realistic or experimental phenomena.
Data availability
Data availability is not applicable to this article as no datasets were generated or analysed during the current study.
References
Karatrantos A, Clarke N, Kröger M (2016) Modeling of polymer structure and conformations in polymer nanocomposites from atomistic to mesoscale: a review. Polym Rev 56(3):385–428
Zhao J, Wu L, Zhan C, Shao Q, Guo Z, Zhang L (2017) Overview of polymer nanocomposites: computer simulation understanding of physical properties. Polymer 133:272–287
Siepmann JI, Karaborni S, Smit B (1993) Simulating the critical behaviour of complex fluids. Nature 365:330–332
Mansfield KF, Theodorou DN (1990) Atomistic simulation of a glassy polymer surface. Macromolecules 23(20):4430–4445
Paul W, Yoon DY, Smith GD (1995) An optimized united atom model for simulations of polymethylene melts. J Chem Phys 103(4):1702–1709
Chen C, Depa P, Sakai VG, Maranas JK, Lynn JW, Peral I, Copley JRD (2006) A comparison of united atom, explicit atom, and coarse-grained simulation models for poly(ethylene oxide). J Chem Phys 124(23):234901
Spyriouni T, Tzoumanekas C, Theodorou D, Müller-Plathe F, Milano G (2007) Coarse-grained and reverse-mapped united-atom simulations of long-chain atactic polystyrene melts: structure, thermodynamic properties, chain conformation, and entanglements. Macromolecules 40(10):3876–3885
Müller-Plathe F (2002) Coarse-graining in polymer simulation: from the atomistic to the mesoscopic scale and back. Chem Phys Chem 3(9):727–824
Ahmad S, Das SK, Puri S (2010) Kinetics of phase separation in fluids: a molecular dynamics study. Phys Rev E 82(4):040107
Graham RS, Olmsted PD (2009) Coarse-grained simulations of flow-induced nucleation in semicrystalline polymers. Phys Rev Lett 103(11):115702
Riniker S, Allison JR, van Gunsteren WF (2012) On developing coarse-grained models for biomolecular simulation: a review. Phys Chem Chem Phys 14(36):12423–12430
Kmiecik S, Gront D, Kolinski M, Wieteska L, Dawid AE, Kolinski A (2016) Coarse-grained protein models and their applications. Chem Rev 116(14):7898–7936
Schwarz KN, Kee TW, Huang DM (2013) Coarse-grained simulations of the solution-phase self-assembly of poly(3-hexylthiophene) nanostructures. Nanoscale 5(5):2017–2027
Srinivas G, Discher DE, Klein ML (2004) Self-assembly and properties of diblock copolymers by coarse-grain molecular dynamics. Nat Mater 3(9):638–644
Jakobsen AF, Mouritsen OG, Besold G (2005) Artifacts in dynamical simulations of coarse-grained model lipid bilayers. J Chem Phys 122(20):204901
Pivkin IV, Karniadakis GE (2006) Coarse-graining limits in open and wall-bounded dissipative particle dynamics systems. J Chem Phys 124(18):184101
Bird RB, Armstrong RC, Hassager O (1987) Dynamics of polymeric liquids. Vol. 1: Fluid mechanics. John Wiley and Sons, New York
Beris AN, Edwards BJ (1994) Thermodynamics of flowing systems with internal microstructure. Oxfold University Press, New York
Groot RD, Warren PB (1997) Dissipative particle dynamics: Bridging the gap between atomistic and mesoscopic simulation. J Chem Phys 107(11):4423–4435
Fermeglia M, Pricl S (2009) Multiscale molecular modeling in nanostructured material design and process system engineering. Comp Chem Eng 33(10):1701–1710
Milano G, Müller-Plathe F (2005) Mapping atomistic simulations to mesoscopic models: a systematic coarse-graining procedure for vinyl polymer chains. J Phys Chem B 109(39):18609–18619
Gooneie A, Schuschnigg S, Holzer C (2017) A review of multiscale computational methods in polymeric materials. Polymers 9(16):1–80
Patnaik SS, Pachter R (2002) A molecular simulations study of the miscibility in binary mixtures of polymers and low molecular weight molecules. Polymer 43(2):415–424
Maiti A, McGrother S (2004) Bead–bead interaction parameters in dissipative particle dynamics: relation to bead-size, solubility parameter, and surface tension. J Chem Phys 120(3):1594–1601
Yang H, Ze-Sheng L, Qian H, Yang Y, Zhang X, Sun C (2004) Molecular dynamics simulation studies of binary blend miscibility of poly(3-hydroxybutyrate) and poly(ethylene oxide). Polymer 45(2):453–457
Paul PD (1978) Polymer blends, vol 1. Academic Press, New York
Utracki LA, Kanial MR (1982) Melt rheology of polymer blends. Polym Eng Sci 22(2):96–114
Utracki LA (1990) Polymer alloys and blends. Hanser Publishers, New York
Flory PJ (1942) Thermodynamics of high polymer solutions. J Chem Phys 10(1):51–61
Huggins ML (1942) Some properties of solutions of long-chain compound. J Phys Chem 46(1):151–158
Flory PJ (1953) Principles of polymer chemistry. Cornell University Press, New York
Ginzburg VV (2005) Influence of nanoparticles on miscibility of polymer blends. A simple theory. Macromolecules 38(6):2362–2367
Coleman MM, Lichkus AM, Painte PC (1989) Thermodynamics of hydrogen bonding in polymer blends. 3. Experimental studies of blends involving poly (4-vinylphenol). Macromolecules 22(2):586–595
Hildebrand JH, Scott RL (1950) The solubility of non-electrolytes. Prentice-Hall, New York
Hansen CM (2007) Hansen solubility parameters: a user’s handbook. CRC Press, Boca Raton
Zhu J, Balieu R, Wan H (2021) The use of solubility parameters and free energy theory for phase behaviour of polymer-modified bitumen: a review. Road Mat Pavement Des 22(4):757–778
González O, Muñoz ME, Santamaría A (2006) Bitumen/polyethylene blends: Using m-LLDPEs to improve stability and viscoelastic properties. Rheol Acta 45(5):603–610
Pérez-Lepe A, Martínez-Boza FJ, Attané P, Gallegos C (2006) Destabilization mechanism of polyethylene-modified bitumen. J App Polym Sci 100(1):260–267
Rojo E, Fernández M, Peña JJ, Peña B, Muñoz ME, Santamaría A (2004) Rheological aspects of blends of metallocene-catalyzed atactic polypropylenes with bitumen. Polym Eng Sci 44(9):1792–1799
Small PA (1953) Some factors affecting the solubility of polymers. J App Chem 3(2):71–80
Merk W, Lichtenthaler RN, Prausnitz JM (1980) Solubilities of fifteen solvents in copolymers of poly (vinyl acetate) and poly (vinyl chloride) from gas-liquid chromatography. Estimation of polymer solubility parameters. J Phys Chem 84(13):1694–1698
Ho RM, Adedeji A, Giles DW, Hajduk DA, Macosko CW, Bates FS (1997) Microstructure of triblock copolymers in asphalt oligomers. J Polym Sci Part B Polym Phys 35(17):2857–2877
Fawcett AH, McNally T (2001) Blends of bitumen with polymers having a styrene component. Polym Eng Sci 41(7):1251–1264
Lindvig T, Michelsen M, Kontogeorgis G (2002) A Flory-Huggins model based on the Hansen solubility parameters. Fluid Phase Equi 203(1):247–260
van Dyk JW, Frisch HL, Wu DT (1985) Solubility, solvency, and solubility parameters. Ind Eng Chem Prod Res Dev 24(3):473–478
Ahmad H, Yaseen M (1979) Application of a chemical group contribution technique for calculating solubility parameters of polymers. Polym Eng Sci 19(12):858–863
Hansen CM (1967) The three dimensional solubility parameter, key to paint component affinities; solvents, plasticizers, polymers and resins. J Paint Technol 39(505):104–117
Stefanis E, Panayiotou C (2008) Prediction of Hansen solubility parameters with a new group-contribution method. Int J Thermophys 29(2):568–585
Mark JE (1996) Physical properties of polymers handbook. AIP Press, New York
Schuld N, Wolf BA (2001) Solvent quality as reflected in concentration- and temperature-dependent Flory-Huggins interaction parameters. J Polym Sci Part B 39(6):651–662
Tambasco M, Lipson JEG, Higgins JS (2006) Blend miscibility and the Flory-Huggins interaction parameter: a critical examination. Macromolecules 39(14):4860–4868
Rubinstein M, Colby RH (2003) Polymer physics. Oxford University Press, New York
Potter CB, Davis MT, Albadarin AB, Walker GM (2018) Investigation of the dependence of the Flory-Huggins interaction parameter on temperature and composition in a drug-polymer system. Macromolecules 15(11):5327–5335
Nedoma AJ, Robertson ML, Wanakule NS, Balsa NP (2008) Measurements of the composition and molecular weight dependence of the Flory-Huggins interaction parameter. Macromolecules 41(15):5773–5779
Russell TP, Hjelm RP Jr, Seeger PA (1990) Temperature dependence of the interaction parameter of polystyrene and poly(methyl methacrylate). Macromolecules 23(3):890–893
Ougizawa T, Inoue T (1986) UCST and LCST behavior in polymer blends and its thermodynamic interpretation. Polymer J 18(7):521–527
Clark EA, Lipson JEG (2012) LCST and UCST behavior in polymer solutions and blends. Polymer 53(2):536–545
Knychała P, Timachova K, Banaszak M, Balsara NP (2017) 50th anniversary perspective: phase behavior of polymer solutions and blends. Macromolecules 50(8):3051–3065
Lefebvre AA, Lee JH, Balsara NP, Hammouda B (2000) Neutron scattering from pressurized polyolefin blends near the limits of metastability. Macromolecules 33(21):7977–7989
Sato T, Endo M, Shiomi T, Imai K (1996) UCST behaviour for high-molecular-weight polymer blends and estimation of segmental parameters from their miscibility. Polymer 37(11):2131–2136
Maruta J, Ougizawa T, Inoue T (1988) LCST-type phase behaviour and extremely slow demixing phenomenon in polymer blends of poly(acrylonitrile-co-styrene) and poly(maleic anhydride-co-styrene). Polymer 29(11):2056–2060
Lewis GN, Randall M, Pitzer KS, Brewer L (1961) Thermodynamics. McGraw-Hill, New York
Newman J, Thomas-Alyea KF (2004) Electrochemical systems. Wiley, Hoboken
Ovejero G, Pérez P, Romero MD, Guzmán I (2007) Solubility and Flory Huggins parameters of SBES, poly(styrene-b-butene/ethylene-b-styrene) triblock copolymer, determined by intrinsic viscosity. Eur Polym J 43(4):1444–1449
Zhao L, Choi P (2002) Measurement of solvent-independent polymer–polymer Flory-Huggins interaction parameters with the use of non-random partitioning solvents in inverse gas chromatography. Polymer 43(25):6677–6681
Metropolis N, Rosenbluth AW, Rosenbluth MN, Teller AH, Teller E (1953) Equation of state calculations by fast computing machines. J Chem Phys 21(6):1087–1092
Alder BJ, Wainwright TE (1957) Phase transition for a hard sphere system. J Chem Phys 27(5):1208–1209
Levitt M, Warshel A (1975) Computer simulation of protein folding. Nature 253(5494):694–698
Padding JT, Briels WJ (2002) Time and length scales of polymer melts studied by coarse-grained molecular dynamics simulations. J Chem Phys 117(2):925–943
Hoogerbrugge PJ, Koelman JMVA (1992) Simulating microscopic hydrodynamic phenomena with dissipative particle dynamics. Europhys Lett 19(3):155–160
Koelman JMVA, Hoogerbrugge PJ (1993) Dynamic simulations of hard-sphere suspensions under steady shear. Europhys Lett 21(3):363–368
Ermack DL, McCammon JA (1978) Brownian dynamics with hydrodynamic interactions. J Chem Phys 69(4):1352–1360
Ameen M (2001) Boundary element analysis: theory and programming. CRC Press, New York
Temam R (2001) Navier-stokes equations: theory and numerical analysis. American Mathematical Soc
Verlet L (1967) Computer “Experiments” on classical fluids. I. Thermodynamical properties of lennard−Jones molecules. Phys Rev 159(1):98–103
Swope WC, Andersen HC, Berens PH, Wilson KR (1982) A computer simulation method for the calculation of equilibrium constants for the formation of physical clusters of molecules: application to small water clusters. J Chem Phys 76(1):637–649
Hockney RW, Eastwood JW (1981) Computer simulation using particles. McGraw-Hill, New York
Beeman D (1976) Some multistep methods for use in molecular dynamics calculations. J Comput Phys 20(2):130–139
Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR (1984) Molecular-dynamics with coupling to an external bath. J Chem Phys 81(8):3684–3690
Nosé S (1984) A molecular dynamics method for simulations in the canonical ensemble. Mol Phys 52(2):255–268
Hoover WG (1985) Canonical dynamics: equilibrium phase-space distributions. Phys Rev A 31(3):1695–1697
Langevin P (1908) On the theory of Brownian motion. C R Acad Sci (Paris) 146:530–533
Smit B, Frenkel D (2002) Understanding molecular simulation, 2nd edn. Academic, New York
Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR (1984) Molecular dynamics with coupling to an external bath. J Chem Phys 81(8):3684–3490
Andersen HC (1980) Molecular dynamics at constant pressure and/or temperature. J Chem Phys 72(4):2384–2393
Sun H (1998) COMPASS: an ab Initio force-field optimized for condensed-phase applications-overview with details on alkane and benzene compounds. J Phys Chem B 102(38):7338–7364
Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M (1983) CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J Comput Chem 4(2):187–217
Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA (1995) A Second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J Am Chem Soc 117(19):5179–5197
Allen MP, Tidesley DJ (1989) Computer simulation of liquids. Clarendon Press, Oxford
Lees AW, Edwards SF (1972) The computer study of transport processes under extreme conditions. J Phys C Solid State Phys 5(15):1921–1929
Kraynik A, Reinelt D (1992) External motions of spatially periodic lattices. Int J Multiph 18(6):1045–1059
Todd BD, Daivis PJ (1998) Nonequilibrium molecular dynamics simulations of planar elongational flow with spatially and temporally periodic boundary conditions. Phys Rev Lett 81(5):1118–1121
Hoover WG, Evans DJ, Hickman RB, Ladd AJC, Ashurst WT, Moran B (1980) Lennard-Jones triple-point bulk and shear viscosities Green-Kubo theory, Hamiltonian mechanics, and nonequilibrium molecular dynamics. Phys Rev A 22(4):1690–1697
Evans DJ, Morriss GP (1990) Statistical mechanics of nonequilibrium liquids. Academic Press, New York
Evans DJ, Morriss GP (1984) Nonlinear-response theory for steady planar Couette flow. Phys Rev A 30(3):1528–1530
Ladd AJC (1984) Equations of motion for non-equilibrium molecular dynamics simulations of viscous flow in molecular fluids. Mol Phys 53(2):459–463
Hoover WG, Hoover CG, Petravic J (2008) Simulation of two- and three-dimensional dense-fluid shear flows via nonequilibrium molecular dynamics: Comparison of time-and-space-averaged stresses from homogeneous Doll’s and Sllod shear algorithms with those from boundary-driven shear. Phys Rev E 78(4):046701
Baig C, Edwards BJ, Keffer DJ, Cochran HD (2005) A proper approach for nonequilibrium molecular dynamics simulations of planar elongational flow. J Chem Phys 122(11):114103
Edwards BJ, Dressler M (2001) A reversible problem in non-equilibrium thermodynamics: Hamiltonian evolution equations for non-equilibrium molecular dynamics simulation. J Non-Newtonian Fluid Mech 96(1):163–175
Delhommelle J, Petravic J, Evans DJ (2003) On the effects of assuming flow profiles in nonequilibrium simulations. J Chem Phys 119(21):11005–11010
Baranyai A (2000) Temperature of nonequilibrium steady-state systems. Phys Rev E 62(5):5989–5997
Li Z, Xiong S, Sievers C, Hu Y, Fan Z, Wei N, Bao H, Chen S, Donadio D, Ala-Nissila T (2019) Influence of thermostatting on nonequilibrium molecular dynamics simulations of heat conduction in solids. J Chem Phys 151(23):234105
Ke Q, Gong X, Liao S, Duan C, Li L (2022) Effects of thermostats/barostats on physical properties of liquids by molecular dynamics simulations. J Mol Liquids 365(1):120116
Anstine DM, Strachan A, Colina CM (2020) Effects of atomistic modeling approach on predicted mechanical properties of glassy polymers via molecular dynamics. Modell Simul Mater Sci Eng 28(2):025006
Kremer K, Grest GS (1990) Dynamics of entangled linear polymer melts: a molecular- dynamics simulation. J Chem Phys 92(8):5057–5086
Marrink SJ, de Vries AH, Mark AE (2004) Coarse grained model for semiquantitative lipid simulations. J Phys Chem B 108(2):750–760
Marrink SJ, Risselada HJ, Yefimov S, Tieleman DP, de Vries AH (2007) The MARTINI force field: coarse grained model for biomolecular simulations. J Phys Chem B 111(27):7812–7824
Grest GS, Kremer K (1986) Molecular dynamics simulation for polymers in the presence of a heat bath. Phys Rev A 33(5):3628–3631
Carmichael SP, Shell MS (2012) A new multiscale algorithm and its application to coarse-grained peptide models for self-assembly. J Phys Chem B 116(29):8383–8393
Goundla Srinivas G, Shelley JC, Nielsen SO, Discher DE, Klein ML (2004) Simulation of Diblock copolymer self-assembly, using a coarse-grain model. J Phys Chem B 108(24):8153–8160
Sun Q, Roland Faller R (2006) Systematic coarse-graining of a polymer blend: polyisoprene and polystyrene. J Chem Theory Comput 2(3):607–615
Sliozberg YR, Mrozek RA, Schieber JD, Kröger M, Lenhart JL, Andzelm JW (2013) Effect of polymer solvent on the mechanical properties of entangled polymer gels: coarse-grained molecular simulation. Polymer 54(10):2555–2564
Harmandaris VA (2014) Quantitative study of equilibrium and non-equilibrium polymer dynamics through systematic hierarchical coarse-graining simulations. Korea-Australia Rheol J 26(1):15–28
Harmandaris VA, Adhikari NP, van der Vegt NFA, Kremer K, Mann BA, Voelkel R, Weiss H, Liew C (2007) Ethylbenzene diffusion in polystyrene: united atom atomistic/coarse grained simulations and experiments. Macromolecules 40(19):7026–7035
Li C, Strachan A (2019) Prediction of PEKK properties related to crystallization by molecular dynamics simulations with a united-atom model. Polymer 174(12):25–32
Heyes DM, Okumura H (2006) Equation of state and structural properties of the Weeks-Chandler-Andersen fluid. J Chem Phys 124(16):164507
Faller R, Kolb A, Müller-Plathe F (1999) Local chain ordering in amorphous polymer melts: Influence of chain stiffness. Phys Chem Chem Phys 1(9):2071–2076
Faller R, Müller-Plathe F, Heuer A (2000) Local reorientation dynamics of semiflexible polymers in the melt. Macromolecules 33(17):6602–6610
Faller R, Müller-Plathe F (2001) Chain stiffness intensifies the reptation characteristics of polymer dynamics in the melt. ChemPhysChem 2(3):180–184
Halverson JD, Grest GS, Grosberg AY, Kremer K (2012) Rheology of ring polymer melts: from linear contaminants to ring-linear blends. Phys Rev Lett 108(3):038301
Composto RJ, Kramer EJ, White DM (1988) Mutual diffusion in the miscible polymer blend polystyrene/poly (xylenyl ether). Macromolecules 21(8):2580–2588
Schlijper AG, Hoogerbrugge PJ, Manke CW (1995) Computer simulation of dilute polymer solutions with the dissipative particle dynamics method. J Rheol 39(3):567–579
Goujon F, Malfreyt P, Tildesley DJ (2009) Mesoscopic simulation of entangled polymer brushes under shear: compression and rheological properties. Macromolecules 42(12):4310–4318
Sandrin D, Wagner D, Sitta CE, Thoma R, Felekyan S, Hermes HE, Janiak C, Amadeu NS, Kühnemuth R, Löwen H, Egelhaaf SU, Seidel CAM (2016) Diffusion of macromolecules in a polymer hydrogel: from microscopic to macroscopic scales. Phys Chem Chem Phys 18(18):12860–12876
Young CD, Zhou Y, Schroeder CM, Sing CE (2021) Dynamics and rheology of ring-linear blend semidilute solutions in extensional flow. Part I: Modeling and molecular simulations. J Rheol 65(4):757–777
Lee HS, Kim WN, Burns CM (1997) Determination of the Flory-Huggins interaction parameter of polystyrene—polybutadiene blends by thermal analysis. Inc J Appl Polym Sci 64(7):1301–1308
Roe RJ, Zin WC (1980) Determination of the polymer-polymer interaction parameter for the polystyrene-polybutadiene pair. Macromolecules 13(5):1221–1228
Chang LL, Woo EM (2003) Surface morphology and Flory-Huggins interaction strength in UCST blend system comprising poly(4-methyl styrene) and isotactic polystyrene. Polymer 44(5):1711–1719
Li J, Ma PL, Favis BD (2002) The role of the blend interface type on morphology in cocontinuous polymer blends. Macromolecules 35(6):2005–2016
Taguet A, Huneault MA, Favis BD (2009) Interface/morphology relationships in polymer blends with thermoplastic starch. Polymer 50(24):5733–5743
Jeon KS, Char K, Walsh DJ, Kim E (2000) Thermodynamics of mixing estimated by equation-of-state parameters in miscible blends of polystyrene and tetramethylbisphenol-A polycarbonate. Polymer 41(8):2839–2845
Han CC, Bauer BJ, Clark JC, Muroga Y, Matsushita Y, Okada M, Tran-cong Q, Chang T, Sanchez IC (1988) Temperature, composition and molecular-weight dependence of the binary interaction parameter of polystyrene/poly(vinyl methyl ether) blends. Polymer 29(11):2002–2014
Shibayama M, Yang H, Stein RS, Han CC (1985) Study of miscibility and critical phenomena of deuterated polystyrene and hydrogenated poly (vinyl methyl ether) by small-angle neutron scattering. Macromolecules 18(11):2179–2187
Londono JD, Wignall GD (1997) The Flory−Huggins interaction parameter in blends of polystyrene and poly(p-methylstyrene) by small-angle neutron scattering. Macromolecules 30(13):3821–3824
Reichart GC, Graessley WW, Register RA, Krishnamoorti R, Lohse DJ (1997) Anomalous attractive interactions in polypropylene blends. Macromolecules 30(10):3036–3041
Londono JD, Narten AH, Wignall GD, Honnell KG, Hsieh ET, Johnson TW, Bates FS (1994) Composition dependence of the interaction parameter in isotopic polymer blends. Macromolecules 27(10):2864–2871
Lefebvre AA, Lee JH, Balsara NP, Hammouda B, Krishnamoorti R, Kumar S (1999) Relationship between internal energy and volume change of mixing in pressurized polymer blends. Macromolecules 32(16):5460–5462
Beiner M, Fytas G, Meier G, Kumar S (2002) Strong isotopic labeling effects on the pressure dependent thermodynamics of polydimethylsiloxane/polyethylmethylsiloxane blends. J Chem Phys 116(3):1185–1192
Lipson JEG, Andrews SS (1992) A Born–Green–Yvon integral equation treatment of a compressible fluid. J Chem Phys 96(2):1426–1434
Gu C, Gu H, Lang M (2013) Molecular Simulation to predict miscibility and phase separation behavior of chitosan/poly (ϵ-caprolactone) binary blends: a comparison with experiments. Macromol Theory Simul 22(7):377–384
Fu Y, Liao L, Yang L, Lan Y, Mei L, Liu Y, Hu S (2013) Molecular dynamics and dissipative particle dynamics simulations for prediction of miscibility in polyethylene terephthalate/polylactide blends. Mol Simul 39(5):415–422
Roths T, Friedrich C, Marth M, Honerkamp J (2002) Dynamics and rheology of the morphology of immiscible polymer blends–on modeling and simulation. Rheol Acta 41(3):211–222
Estridge CE, Jayaraman A (2015) Diblock copolymer grafted particles as compatibilizers for immiscible binary homopolymer blends. ACS Macro Lett 4(2):155–159
Jones JE (1924) On the determination of molecular fields.—I. From the variation of the viscosity of a gas with temperature. Proc R Soc London Ser A 106(738):441–462
Lennard-Jones JE (1931) Cohesion. Proc Phys Soc 43(5):461–482
Kozuch DJ, Zhang W, Milner ST (2016) Predicting the Flory-Huggins χ parameter for polymers with stiffness mismatch from molecular dynamics simulations. Polymers 8(6):241
Luo Z, Jiang J (2010) Molecular dynamics and dissipative particle dynamics simulations for the miscibility of poly (ethylene oxide)/poly (vinyl chloride) blends. Polymer 51(1):291–299
Spyriouni T, Vergelati C (2001) A molecular modeling study of binary blend compatibility of polyamide 6 and poly(vinyl acetate) with different degrees of hydrolysis: an atomistic and mesoscopic approach. Macromolecules 34(15):5306–5316
Chuang HK, Han CD (1984) Rheological behavior of polymer blends. J App Polym 29(6):2205–2229
Zhang ZL, Zhang HD, Yang YL, Vinckier I, Laun HM (2001) Rheology and morphology of phase-separating polymer blends. Macromolecules 34(5):1416–1429
Hamad K, Kaseem M, Deri F (2010) Rheological and mechanical properties of poly (lactic acid)/polystyrene polymer blend. Polym Bull 65(5):509–519
Huang LH, Wu CH, Hua CC, Huang TJ (2020) Multiscale simulations of coupled composition-stress-morphology of binary polymer blend. Polymer 193:122366
Peng W, Ranganathan R, Keblinski P, Ozisik R (2017) Viscoelastic and dynamic properties of well-mixed and phase-separated binary polymer blends: a molecular dynamics simulation study. Macromolecules 50(16):6293–6302
Iza M, Bousmina M, Jérôme R (2001) Rheology of compatibilized immiscible viscoelastic polymer blends. Rheol Acta 40(1):10–22
Huang LH, Hua CC (2022) Predictive mesoscale simulation of flow-induced blend morphology, interfacial relaxation, and linear viscoelasticity of polymer-elastomer blends. Macromolecules 55(17):7353–7367
Wang J, Cao C, Chen X, Ren S, Yu D, Chen X (2019) Extensional-shear coupled flow-induced morphology and phase evolution of polypropylene/ultrahigh molecular weight polyethylene blends: Dissipative particle dynamics simulations and experimental studies. Polymer 169(15):36–45
Hajizadeh E, Todd BD, Daivis PJ (2015) A molecular dynamics investigation of the planar elongational rheology of chemically identical dendrimer-linear polymer blends. J Chem Phys 142(17):174911
Bouzid L, Hiadsi S, Bensaid MO, Foudad FZ (2018) Molecular dynamics simulation studies of the miscibility and thermal properties of PMMA/PS polymer blend. Chinese J Phys 56(6):3012–3019
Lee CK, Pao CW, Chu CW (2011) Multiscale molecular simulations of the nanoscale morphologies of P3HT:PCBM blends for bulk heterojunction organic photovoltaic cells. Energy Environ Sci 4(10):4124–4132
Billeter M, Guntert P, Luginbuhl P, Wuthrich K (1996) Hydration and DNA recognition by homeodomains. Cell 85(7):1057–1065
Ganazzoli F, Raffaini G (2005) Computer simulation of polypeptide adsorption on model biomaterials. Phys Chem Chem Phys 7(21):3651–3663
Harmandaris VA, Angelopoulou D, Mavrantzas VG, Theodorou DN (2002) Atomistic molecular dynamics simulation of diffusion in binary liquid n-alkane mixtures. J Chem Phys 116(17):7656–7665
Gestoso P, Brisson J (2003) Investigation of the effect of chain rigidity on orientation of polymer blends: the case of poly (vinyl phenol)/poly (ethylene terephthalate) blends. Polymer 44(25):7765–7776
Faller R (2004) Correlation of static and dynamic inhomogeneities in polymer mixtures: a computer simulation of polyisoprene and polystyrene. Macromolecules 37(3):1095–1101
Deng S, Payyappillyb SS, Huanga Y, Liu H (2016) Multiscale simulation of shear-induced mechanical anisotropy of binary polymer blends. RSC Adv 6(48):41734–41742
O’Connor TC, Ge T, Grest GS (2022) Composite entanglement topology and extensional rheology of symmetric ring-linear polymer blends. J Rheol 66(1):49–65
Kröger M (2005) Shortest multiple disconnected path for the analysis of entanglements in two-and three-dimensional polymeric systems. Comput Phys Comm 168(3):209–232
Jeong SH, Kim JM, Yoon J, Tzoumanekas C, Kröger M, Baig C (2016) Influence of molecular architecture on the entanglement network: topological analysis of linear, long- and short-chain branched polyethylene melts via Monte Carlo simulations. Soft Matter 12(16):3770–3786
Ge T, Grest GS, Robbins MO (2013) Structure and strength at immiscible polymer interfaces. ACS Macro Lett 2(10):882–886
Salahshoori I, Jorabchi MN, Valizadeh K, Yazdanbakhsh A, Bateni A, Wohlrab S (2022) A deep insight of solubility behavior, mechanical quantum, thermodynamic, and mechanical properties of Pebax-1657 polymer blends with various types of vinyl polymers: a mechanical quantum and molecular dynamics simulation study. J Mol Liq 363:119793
Wei M, Xu P, Yuan Y, Tian X, Sun J, Lin J (2018) Molecular dynamics simulation on the mechanical properties of natural-rubber-graft-rigid-polymer/rigid-polymer systems. Phys Chem Chem Phys 20(12):8228–8240
Li J, Jin S, Lan G, Chen S, Li L (2018) Molecular dynamics simulations on miscibility, glass transition temperature and mechanical properties of PMMA/DBP binary system. J Mol Graph 84:182–188
Watt JP, Davies GF, O’Connell RJ (1976) The elastic properties of composite materials. Rev Geophys Space Phys 14(4):541–563
Zhao X, Deng S, Huang Y, Liu H, Hu Y (2011) Simulation of morphologies and mechanical properties of A/B polymer blend film. Chinese J Chem Eng 19(4):549–557
Acknowledgements
This research was supported by the National Research Foundation of Korea (NRF) (RS-2022-00165711) and Institute of Information & Communications Technology Planning & Evaluation (IITP) (2020-0-01791) grant funded by the Korea government (MSIT).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jeong, S. Investigation of intrinsic characteristics of polymer blends via molecular simulation: a review. Korea-Aust. Rheol. J. 35, 249–266 (2023). https://doi.org/10.1007/s13367-023-00076-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13367-023-00076-9