
Abstract
In the present work, the high-temperature decarburization of the austenitic stainless steel S304HCu in Ar–4%H2–xH2O at temperatures between 1000 and 1150 °C was investigated. The focus was on determining parameters that influence the decarburization rate during heat treatment. Thermogravimetric experiments were performed with simultaneous measurements of CO release from the sample using a mass spectrometer. The variation of the experimental parameters included the composition of the atmosphere during pre-oxidation and decarburization, the temperature and the sample thickness. The results indicate that the loss of carbon from the specimen occurs faster through reaction of Cr2O3 with C from the steel compared to the reaction of C at the steel surface with H2O, provided that in the former case CO can easily evaporate. The overall decarburization process is governed by competing reactions; the destruction of chromium oxide by C and the formation of oxide scale by water vapour. The relative rates of these two reactions determines whether decarburization will occur or whether formation of a protective oxide stops the release of CO, the latter being promoted at lower temperatures and high water vapour contents. A dense, sufficiently thick chromia scale retards the onset of decarburization but does eventually not stop the carbon loss. Indications were found that the destruction of a pre-formed chromia scale by carbon involves a two-step process whereby presence of hydrogen is important.
Similar content being viewed by others


Introduction
Decarburization is a loss of carbon atoms from the surface of the work pieces which commonly occurs when carbon or low alloy steel is heated in an environment where oxygen is present, leading to oxidation and loss of carbon. Oxidation and decarburization are frequently found to occur simultaneously in the hot rolling or annealing processes [1]. In most cases decarburization of carbon or low alloy steel, which commonly occurs during air heat treatment at temperatures above approximately 700 °C, is an undesired phenomenon because it will adversely affect the surface mechanical properties of the steel [2].
In other applications such as electrical steels and enameling, decarburization is intentionally induced during annealing [3, 4]. In these cases, the heat treatment is commonly carried out in an atmosphere with low oxygen partial pressure to prevent surface oxidation, producing steel strip with very low-carbon concentration. Contrary to carburizing or nitriding, annealing of electrical steel relies on the removal of carbon, not just from the surface but from the total volume of the steel [5]. Also in the case of high alloyed materials such as austenitic steels, a reduced (surface) carbon content may be beneficial because it can prevent sensitization and thus improve corrosion resistance of such materials [3,4,5].
Basically, during heat treatment of low alloy steel in an oxidizing atmosphere such as air above approximately 570 °C when they are reheated for hot working or for heat treatment [6, 7], a scale of iron oxide forms and grows. At the scale—alloy interface, carbon interacts with the scale to form carbon monoxide by the reaction (1).
This reaction can only proceed if the reaction product carbon monoxide can escape through the scale. In general, porous scales are produced, particularly under industrial conditions, and removal of carbon monoxide is not a problem. It is important to note that all these processes are thermally activated and will be accelerated by an increase in temperature.
Boggs and Kachik [8] showed that decarburization of iron carbon alloys can even occur at temperatures as low as 500 °C. In that case, in Eq. (1) the equilibrium between C and Fe3O4 rather than FeO is relevant. The authors found the alloys to decarburize when oxidized in oxygen at 10 Torr, but do not decarburize when oxidized at oxygen at 700 Torr. This effect of oxygen pressure can be explained if it is assumed that pores or faults are left at the oxide grain boundaries. Carbon from the substrate reacts with CO2 in the pore atmosphere to form CO. Thus, the pores provide paths for the escape of carbon oxides into the atmosphere. When the external oxygen pressure is 10 Torr, oxygen entering the pores reacts with CO to form CO2. This mechanism maintains a CO–CO2 atmosphere of low oxygen activity within the pores, keeping them open. However, at the higher pressure, 700 Torr, oxygen is able to enter the pores and be incorporated in the iron-oxide lattice. This causes the growth of fresh oxide in the pores, blocking them and preventing the escape of carbon oxides.
When high alloyed materials such as austenitic stainless steels or nickel-based alloys are heat treated in air at high temperatures, decarburization is commonly not observed. This is mainly attributed to the growth of chromia-based oxide scales forming on the material surface, which offer far better protection against access of oxygen than iron-based oxides. In analogy to the discussion above, the important issue for decarburization to occur would be the carbon and oxygen activity at the scale alloy interface, governed by reaction (2)
\(K_{2}\) is the equilibrium constant for reaction (2), \(p{\text{CO}}\) is the equilibrium carbon monoxide partial pressure at the scale alloy interface, \(a_{{\text{C}}}\) and \(a_{{{\text{Cr}}}}\) are the alloy carbon and Cr activity, respectively.
Due to the very high thermodynamic stability of chromia, the equilibrium CO partial pressure of Eq. (2) is orders of magnitude smaller than that of Eq. (1) (see quantitative data in next chapter) and thus the thermodynamic driving force for reaction 2 is much smaller. Additionally, the chromia-based surface scales do not exhibit the extensive voidage and cracking commonly seen for the Fe-based oxides; and thus, the formed CO cannot easily escape.
In practical applications, decarburization of the mentioned high alloyed materials has been observed after heat treatment in “non-oxidizing,” protective hydrogen-based atmospheres, the latter being used to prevent or at least minimize surface oxidation such as, e.g., during bright annealing processes [9]. Similar decarburization effects have been found during sintering of high alloyed materials in hydrogen or vacuum [10] and also during fluxless vacuum brazing of austenitic steels [11].
As far as known to the authors, hardly any systematic quantitative studies about the parameters affecting decarburization occurring during these practically relevant heat treatment processes of the mentioned high alloyed materials are available. However, in the mid-eighties of the twentieth century, extensive studies concerning decarburization processes of mainly nickel-based alloys during high temperature exposure in so called high-temperature reactor (HTR) helium were carried out [12, 13]. In the HTR helium, which contains impurities of H2, H2O, CO and CH4 in the microbar range, nickel-based alloys such as INCONEL 617 and HASTELLOY X were found to decarburize at and above approximately 950 °C, depending on the detailed gas composition. In HTR helium typically containing (in microbar) 500 H2, 15 CO, 20 CH4 and 1 H2O, decarburization may occur by direct reaction of C with water vapour (forming CO and H2) but even under conditions where chromia surface scale formation was found, decarburization occurred which was attributed to reaction of carbon with the chromia surface scale (reaction 2). Apparently, the reaction resulted in damage of the protective surface scale thus providing sufficient paths along which the CO could escape into the atmosphere. This mechanism was confirmed by the fact that even during exposure for several thousands of hours at 950 °C, decarburization could be prevented by increasing the CO content in the gas to a value (typically 50 microbar) which is higher than the equilibrium CO partial pressure of Eq. (2). More recently other authors, e.g., Cabet et al. [14, 15] and Raghavendra et al. [16] carried out investigations on the mechanisms of decarburization under HTR conditions. In the paper of Cabet et al. [14] it was found, that reduced pCO in the He-based test gas led to the consumption of dissolved carbon in the studied nickel-based alloys (Alloy 617 and Alloy 230) through the reaction of chromia with carbon from the alloy to produce CO(g) and metallic chromium. The authors found, for example, that Alloy 617 lost about 50% of its initial carbon during a 500 h exposure at 950 °C in the flowing impure helium. Similar conclusions were achieved by Raghavendra et al. [16] who studied the microstructure and surface stability of two experimental W-rich Ni-based alloys at 950 °C in an impure He environment containing only CO and CO2 as impurities. The protective surface chromium oxide was reduced by the solid-solution carbon present in the substrate, thus leading to a chromia-assisted decarburization reaction. The authors showed that the formation of Cr2O3 was the rate-limiting step in the proposed decarburization reaction and that for the pre-oxidized alloys, chromia-assisted decarburization reactions were slower for an alloy containing carbides compared to the alloy with carbon in solid solution only. An additional observation was that, the presence of even a thin layer (< 1 µm) of Al2O3 between the substrate and surface Cr2O3 oxide inhibited decarburization and/or Cr2O3 loss.
An important finding described in [12, 13] was, that decarburization of a nickel-based alloy such as INCONEL 617 in HTR Helium could not only be stopped by adding CO to the test gas but also by significantly increasing the water vapour content (to e.g., 100 ppm at 950 °C). This finding seems at first sight to be surprising because a higher water vapour content provides an additional source of oxygen and would therefore be expected to increase the tendency for decarburization. However, gas chromatographical analyses of the CO evolution during increasing and decreasing the water vapour content in a He-500 ppm H2 gas provided insight into the prevailing mechanisms. In the Cr2O3 surface scale, even in the isothermal case, minor amounts of cracks or pores are continuously formed (e.g., due to growth stresses in combination with the reaction of C reacting with the scale [17] through which the CO can escape. If the H2O content is very low (e.g., 1 ppm), the oxidation rate by H2O is too small compared to the oxide reduction rate by carbon [Eq. (2)] for scale imperfections to be healed. Therefore, the carbon loss cannot be stopped by a forming or a pre-formed Cr2O3 scale. In atmospheres with high H2O levels (e.g., 100 ppm, at 950 °C and an H2 content of 500 ppm) the oxidation rate is high compared with the oxide reduction rate, resulting in a rapid healing [18] of the imperfections so that the surface is sealed. Consequently, the CO cannot escape, and the oxide destruction and decarburization are stopped.
These results show, that, under conditions where the equilibrium oxygen partial pressure in the gas is sufficiently high for chromia scales to be formed, the question whether decarburization of the high alloyed materials occurs, cannot be derived from thermodynamic considerations related to, e.g., the equilibrium oxygen partial pressure and/or carbon activity. The scale formation is a dynamic process in which reaction of carbon at the oxide-alloy interface, crack/void formation and crack/void healing are competing. The kinetics of these reactions is governed by absolute contents of water vapour and hydrogen rather than the equilibrium oxygen partial pressure. It is therefore hardly possible to quantitatively transfer the findings from the studies in HTR helium with the very low hydrogen and water vapour partial pressures to conditions commonly encountered in heat treatment practice, i.e., a hydrogen rich gas with a defined dew point. At a total pressure of one bar, a water vapour content of 1 ppm, typically for HTR helium, corresponds to a dew point of − 80 °C. This is hardly economically achievable in most practical applications.
In the present study, the decarburization behavior of a selected high alloyed material, here the stainless steel S304HCu, is investigated in a range of atmospheres which contain substantially higher hydrogen and water vapour contents than those used in HTR helium [19]. As base gas Ar–4% H2 instead of pure H2 was selected to avoid safety considerations in carrying out the thermogravimetric studies. Main emphasis was put on measuring decarburization kinetics under conditions where the equilibrium oxygen partial pressure of the gas allows formation of chromia-based surface scales. The time and temperature dependence of the decarburization and oxidation reactions was measured using thermogravimetry with parallel analysis of the gas directly after the reacting sample using mass spectrometry.
In the following, after a brief presentation of the experimental set-up a more extensive description of the theoretical background for decarburization providing quantitative data, will be shown. This is important because it provides the thermodynamic data which for instance illustrate the exact conditions (gas, temperature) under which chromia formation is possible, etc. Subsequently after presenting the experimental details, results of the gravimetric and gas analyses for various experimental conditions are provided and discussed.
Experimental Procedures
Table 1 gives the detailed chemical composition of the studied steel S304HCu analyzed by inductively coupled plasma–optical emission spectroscopy (ICP-OES) and infrared (IR) analysis. The material was supplied by Mannesmann DMV Stainless in form of seamless stainless steel tubes. The final production step was solution annealing at 1150 °C. For the planned decarburization tests, specimens of 20 mm × 10 mm × x mm (specimens with various thickness were tested x = 1 and 3 mm) were cut from the supplied tubes and subsequently ground down to 1200 grit surface finish with SiC abrasive paper. Prior to the oxidation tests, the samples were degreased in an ultrasonic bath with ethanol. The isothermal experiments were carried out in air, Ar–4%H2–x ppm H2O and Ar–x ppm H2O using a microbalance (Setaram TGA 16) connected to a mass spectrometer (QuadStar, QMS 200), a thermostat (Julabo Fw95SL) and a moisture measuring device (SYSTECH INSTRUMENTS Model 510).
Most of the decarburization experiments were preceded by a pre-oxidation carried out in the same gas as the subsequent decarburization treatment. In that case the pre-oxidation and subsequent decarburization occurred continuously without intermediate cooling of the specimen. Thus, the mass gain during heating from pre-oxidation temperature to decarburization temperature was partly related to the buoyancy effect and not only to the weight gain associated with the formation of oxide. In the case of “non-continuous” exposure, when pre-oxidation and decarburization were carried out separately, the weight gain curves were appropriately corrected by subtracting the buoyancy effect.
With the mass spectrometer connected to the microbalance the intensity of the most significant evolving gaseous species, CO evaporating during the present experiments was measured. The CO peak started to be detectable during specimen heating after exceeding a given temperature and indicated the start of the decarburization process. The mass spectra were not explicitly quantified. However, the parameters used during the analysis were kept the same for all experiments so that the relative amounts of CO in the various experiments could directly be compared.
After the exposures, selected specimens were analyzed using optical metallography and SEM.
To visualize the grain shape and size, the distribution of precipitates and inclusions that are present in the alloy, selected specimens were etched with various etchants, i.e., V2A etchant, NH3-solution or 5% H2SO4-solution.
For measuring the carbon profile in the decarburized specimens glow discharge optical emission spectroscopy (GDOES) was used (Horiba Jobin Yvon, HR Profiler).
Selected specimens were after exposure analyzed in respect to bulk carbon content using combustion/infrared analysis.
Theoretical Background
When considering decarburization of high alloyed metallic materials (e.g., austenitic steels, Ni-based alloys) in H2/H2O-based atmospheres the following decarburizing reactions may be considered:
The thermodynamic driving force for the decarburization process will be governed by the partial pressure of CH4 [Eq. (4)] and CO in equilibrium with the H2 [and H2O, Eq. (5)] partial pressure in the gas and the C activity in the alloy [12, 20].
Considering a test gas on the basis of Ar–4 vol% H2, the equilibrium CH4 partial pressure can be derived from
here \(K_{4}\) is the equilibrium constant of reaction (4), \(a_{C}\) the carbon activity at the metal surface and \(pH_{2}\) the hydrogen partial pressure.
The calculated activities of species (using FACTSAGE plus data base FACT) assuming equilibrium between the mentioned test gas and a typical C activity (a = 10–3) for high alloyed materials [21] in the temperature range 500 to 1100 °C show that the equilibrium value of pCH4 is quite low and decreases with increasing temperature from 3.5 × 10–6 bar at 500 °C to 7.8 × 10–9 bar at 1100 °C. Thus, at temperatures at which reaction kinetics would be expected to be sufficiently fast, the thermodynamic driving force for the decarburization due to methane formation is very low. Additionally, it should be considered that the diffusivity of hydrogen in austenitic steels and nickel base alloys is substantially larger than that of carbon [22, 23]. This has as a result that hydrogen diffuses far more rapidly into the material than carbon diffuses outward. Consequently, the reaction (4) would tend to occur within the material rather than at the surface. For these reasons, in addition to the fact that the kinetics of reaction (4) are substantially slower than that of reaction (5) [20] a sufficiently rapid decarburization based on reaction (4) seems not to be possible.
Using Eq. 5 the partial pressure of CO in equilibrium with the used Ar–4%H2 atmosphere, the alloy carbon activity of 10–3 and an arbitrarily chosen water vapour partial pressure of 10–4 bar (i.e., 100 ppm) can be derived from
here \(K_{5}\) is the equilibrium constant for reaction (5), \(a_{{\text{C}}}\) the carbon activity, pH2 the hydrogen partial pressure and \(p_{{{\text{H}}_{2} {\text{O}}}}\) the water vapour partial pressure.
Using the same thermodynamic data base as mentioned above, calculated values for pCO are 5.2 × 10–8 bar at 500 °C and 5.3 × 10–4 bar at 1100 °C. It is apparent that pCO increases with increasing temperature and reaches at higher temperatures quite high values. For this reason, in combination with the rapid kinetics [20], reaction (5) seems to be more suitable than reaction (4) for obtaining rapid decarburization rates.
From Eq. (7) it is apparent that for a given \(p{\text{H}}_{2}\) and \(a_{{\text{C}}}\), the equilibrium value for pCO increases with increasing \(p{\text{H}}_{2} {\text{O}}\), i.e., high water vapour contents should tend to increase the driving force for decarburization. However, it should be borne in mind that for the considered high alloyed austenitic steel, high water vapour contents will increase the equilibrium oxygen partial pressure in the gas and thus the tendency for formation of surface oxide scales, here mainly consisting of chromia.
\(K_{8}\) is the equilibrium constant for reaction (8), \(p{\text{O}}_{2}\) the oxygen partial pressure, \(p{\text{H}}_{2}\) the hydrogen partial pressure and \(p{\text{H}}_{2} {\text{O}}\) the water vapour partial pressure. If the \(p{\text{O}}_{2}\) given by reaction (8) is higher than the dissociation pressure of Cr oxide in equilibrium with the respective Cr activity in the alloy, then Cr is expected to become oxidized. The mentioned dissociation pressure may be derived from the reaction (10)
\(K_{10}\) is the equilibrium constant for reaction (10), \(p{\text{O}}_{2}\) the oxygen partial pressure (i.e., the dissociation pressure of Cr2O3). It is apparent from reaction (11) that the dissociation pressure increases with decreasing activity of Cr in the alloy.
Table 2 shows the amount of H2O (in ppm) in the Ar–4%H2 gas necessary for Cr to become oxidized at a given temperature. At this gas composition the pO2 given by reaction (8) equals the dissociation pressure of Cr oxide [Eq. (10)]. In the calculations, it was assumed that the Cr activity equals 0.4 and the base gas consists of Ar–4 vol% H2. The data in Table 2 illustrate that with increasing temperature higher contents of water vapour are required to oxidize Cr.
Equation (7) illustrates that increasing the water vapour content increases the driving force for decarburization. However, for a given value of pH2, the alloy will tend to form surface chromia scales if the water vapour content exceeds a certain value which may be derived from Table 2. If surface chromia scale formation occurs, direct access of water vapour to the alloy surface is hampered. Additionally, CO which would be formed at the alloy surface cannot freely “escape” to the atmosphere and thus the decarburization process would finally be stopped, i.e., the alloy would become “passivated.”
In reference [24, 25] it was shown that in presence of a chromia scale during exposure of the above-mentioned metallic materials in (Ar–)H2–H2O base gas mixtures the additional decarburization reaction (2) may occur.
Table 3 shows the activities for Cr and C and the resulting equilibrium pCO values, given by Eq. (3), in the temperature range 550 to 1100 °C for S304HCu. The table also shows the corresponding values for a carbon steel (Fe–1%C), here for Fe and C activities. The respective Fe, Cr and C activity values in Table 3 were calculated using ThermoCalc and the database TCFE6.
Strictly spoken, reaction (2) could also be written with CO2 instead of CO but at the very low pO2 at the scale alloy interface the CO2 value is orders of magnitudes lower than that of CO, e.g., at 1000 °C the equilibrium value of pCO equals 2.7 × 10–4 bar; whereas, the equilibrium pCO2 equals 3.2 × 10−7bar. This is also the case in the H2/H2O gas used; the pO2 is higher than that at the scale-alloy interface but still the equilibrium CO2 value is extremely small, i.e., insignificant compared to that of CO.
The data in Table 3 illustrate that the partial pressures of CO produced by reaction between Cr2O3 and C increase substantially with increasing temperature. In the case of carbon steel possessing high C activity extremely high CO pressures are developed at high temperatures [26].
From Eq. (2) it is apparent that this reaction proceeds at the chromia-alloy interface [27]. It incorporates a “destruction” of the chromia scale by carbon and consequently a removal of carbon from the alloy. Not necessary to explain that ongoing decarburization via this reaction would require that the CO can freely “escape” into the gas, e.g., via microcracks in the oxide scale. Whether this is the case for the prevailing experimental condition depends on the rate of oxide formation at the surface via the reaction
and the rate of the decarburizing reaction (2). If the rate of the latter reaction is faster than that of the oxidizing reaction (12) the oxide scale will continue to be “destroyed” and thus the decarburization process will continue. If the opposite is the case, then surface oxide formation will finally dominate and the alloy will become “passivated,” i.e., the decarburization process ceases.
Figure 1 illustrates qualitatively the “window” for decarburization for austenitic steels such as S304HCu in an (Ar–)H2–H2O base gas mixture. Considering reaction (5), the driving force for decarburization increases with increasing water vapour content. However, above a critical water vapour partial pressure (pH2O), depending on the hydrogen partial pressure in the gas and the Cr activity in the alloy, chromia surface scale formation is expected [Eq. (12), Table 2]. The chromia scale may be “destroyed” by the decarburizing reaction (2) whereby the thermodynamic driving force (proportional to pCO) increases with increasing temperature and increasing C activity in the alloy. Decarburization may thus still occur in spite of the fact that a chromia surface scale can be formed. This will proceed as long as the rate of oxidation at the metal surface by reaction (12) is lower than the rate of oxide “destruction” by reaction (2). So, decarburization will eventually stop above a critical water vapour content in the gas. As seen from Table 3, the pCO in equilibrium with the oxide and the alloy carbon activity decreases with decreasing temperature. For instance for alloy S304HCu the equilibrium value becomes less than 1 µbar beneath temperatures of 800 °C. Thus the thermodynamic driving force for reaction (2) is then extremely small and thus the oxidation reaction (12) will always dominate. Consequently, at these low temperatures decarburization will not occur. In fact, the pO2 required to oxidize Cr, decreases with decreasing temperature. So, the thermodynamic driving force for oxidation increases with decreasing temperature. The thermodynamic driving force for oxide reduction by carbon decreases with decreasing temperature.

Schematic illustrating competition between oxide scale formation and decarburization in (Ar–)H2–H2O
Results and Discussion
In order to verify the validity of the decarburization mechanisms described in previous chapter, and to quantitatively estimate the parameters affecting the conditions under which decarburization occurs and how the various experimental parameters are affecting the decarburization rates, thermogravimetry experiments were performed in Ar–H2/H2O or Ar–H2O atmospheres. The experiments included a pre-oxidation step at relatively low temperatures (700–900 °C) and a de-carburization step at higher temperatures (1000–1150 °C) without intermediate specimen cooling. Using this procedure allowed investigation of the decarburization reaction kinetics due to reduction of Cr oxide according to reaction (2), depending on the water vapour content in the gas.
Decarburization with Pre-oxidation in Ar–H2/H2O and Ar–H2O
Effect of H2O Content in the Decarburization Atmosphere
Figure 2a shows weight changes as a function of “pre-oxidation/decarburization” time for S304 HCu in Ar–4%H2–x ppm H2O. The pre-oxidation period at 700 °C was 20 h; after this time the specimens were heated up to the decarburization temperature 1050 °C (heating rate 1.67 K/min). The water vapour content in the prevailing tests varied from 10 to 300 ppm. During the pre-oxidation treatment at 700 °C a small weight gain was observed, which showed a near parabolic time dependence as one would expect for growth of a thin chromia-based oxide layer on the surface [18].

Weight changes and temperature as function of time for S304 HCu specimens during pre-oxidation at 700 °C and subsequently decarburization at 1050 °C in a Ar–4%H2–x ppm H2O and b Ar–x ppm H2O, specimen thickness 1 mm
Figure 2a also shows that, in agreement with literature data for chromia growth [28], the oxidation rate is not substantially affected by the variations in water vapour content between 10 and 300 ppm. Assuming the scale to consist of pure chromia, the weight gain of 0.03 mg/cm2 after 20 h corresponds to a thickness of approximately 0.18 µm.
During heating from the pre-oxidation to the decarburization temperature in Ar–H2/H2O, faster oxidation of the specimen surfaces occurs manifested by an initial increase in mass gain. Subsequently, the specimen exposed in the 10 ppm H2O gas exhibits a continuous mass loss till the end of exposure. In the 300 ppm H2O gas a continuous weight gain occurs, although with a slower rate than during heat up. In the 100 ppm H2O gas in intermediate case occurs; after the initial weight gain a weight loss occurs whereas later the weight again increases. Generally, specimens exposed in the gases with lower water vapour content (10 ppm) show higher net weight loss than specimens decarburized in the gases with higher water vapour content. In the case of specimens decarburized in the gas with high water vapour pressure of 300 ppm no net weight loss was observed.
Figure 2b shows weight changes as function of “pre-oxidation/decarburization” time for S304 HCu in Ar–x ppm H2O, i.e., test gases which, in contrary to the previous series of experiments, do not contain added hydrogen. The results show that specimens exposed in gases with lower water vapour content (10 ppm) show smaller weight gains than specimens exposed in gases with higher water vapour content. In the gases with 100 and 300 ppm H2O a continuous weight gain occurs. For 10 ppm H2O an initial weight gain is followed by a step with virtually no weight change between 26 and 34 h; subsequently a weight gain again occurs.
Figure 3 shows for the experiments in the Ar–H2–H2O and Ar–H2O gases with the highest and lowest water vapour contents the CO values measured by mass spectrometry. In all cases CO formation is observed just before reaching the maximum temperature of 1050 °C. It is important to mention that in all experiments described in the present study, no indication for methane or CO2 formation was found thus showing that neither decarburization by direct reaction with hydrogen, nor formation of CO2 as a result of the carbon-oxide reaction contribute to the overall decarburization process, in agreement with the considerations in “Theoretical Background” section.

CO intensities versus time measured by mass spectrometer for S304 HCu specimens during pre-oxidation at 700 °C and subsequently decarburization at 1050 °C in a Ar–4%H2–x ppm H2O and b Ar–x ppm H2O, specimen thickness 1 mm
During the isothermal exposure the CO production gradually declines whereby the totally produced amount of CO is larger for the experiments with the lower water vapour content which indicates that the CO formation is not caused by a direct reaction of H2O with C [reaction (5)]. Also, the amount of evolved CO is much larger in the atmosphere with compared to that without 4% hydrogen. These results clearly show that the total measured weight change shown in Fig. 2 is a combination of weight gain due to oxidation and a weight loss due to decarburization. Based on the CO analyses it can be derived that the differences in behavior in respect to weight loss are caused by several processes running in parallel. For higher water vapour content decarburization rapidly decreases after an “initial peak” (Fig. 3a, b). Then, the rate of chromium oxide formation is faster than chromium oxide reduction by carbon [Eq. (2)] leading to the formation of a passive layer that prevents further escape of CO.
The results show a clear difference in respect to weight change between these two atmospheres thus revealing a significant influence of hydrogen partial pressure on the decarburization process. The mechanisms for this finding will be discussed later.
Effect of Decarburization Temperature
The influence of the decarburization temperature is shown in Figs. 4 and 5 which compare specimens exposed at various temperatures but in the same test gas, i.e., Ar–4% H2–100 ppm H2O using the pre-oxidation treatment as that used in Fig. 2. The 1050 °C curve shown in Fig. 4a is identical to that in Fig. 2a. For both tests a weight gain occurs upon heating from 700 °C to the decarburization temperature of 1000 or 1050 °C. However, whereas at/near the temperature of 1050 °C a weight loss starts to occur, a continuous weight gain is seen in case of the 1000 °C exposure, although the slope of the curve changes approximately upon reaching the maximum temperature.

a Weight changes as function of time during “pre-oxidation/decarburization” and b CO intensities versus time measured by mass spectrometry during exposure of 1 mm thick S304 HCu specimens in Ar–4%H2–100 ppm H2O at 700 °C pre-oxidation and subsequently at 1000 and 1050 °C decarburization. Temperature time plots are indicated by dashed lines in both figures

Weight changes as function of time during “pre-oxidation/decarburization” of 1 mm thick S304 HCu specimens in Ar–4%H2–100 ppm H2O at 700 °C pre-oxidation for 2 h and subsequently decarburization at 1050 and 1100 °C
The mass change measured during 1000 °C decarburization (Fig. 4a) is increasing continuously with time. However, mass spectrometry measurements of CO in Fig. 4b show that, in spite of the continuous weight gain, a minor decarburization occurs between approximately 30 and 40 h total exposure time. During exposure of 1050 °C enhanced oxidation is observed during heating similar to 1000 °C. However, after heating a strong CO release occurs (Fig. 4b) indicating rapid decarburization accompanied by a significant decrease in the mass change (Fig. 4a). After about 30 h total exposure the mass loss rate decreases and at longer time a positive mass change is observed. This can be related to decreased availability of C at the surface for the decarburization process as will be discussed in the section below.
Also Fig. 5 shows a clear effect of temperature on the decarburization rate. The weight change as function of time for specimens during pre-oxidation (here only 2 h) and decarburization at two different temperatures 1050 and 1100 °C (pre-oxidation 2 h at 700 °C, decarburization 20 h in Ar–4%H2–100 ppm H2O) are virtually identical up to approximately 6 h. Subsequently the weight of the specimen decarburized at 1100 °C sharply decreases indicating further decarburization up to about − 0.3 mg/cm2; whereas, the decarburization for the specimen decarburized at 1050 °C seems to be virtually stopped at about − 0.15 mg/cm2. After an initial strong weight decrease (up to approximately 15 h) both curves show a slight weight increase expectedly due to ongoing oxide scale formation whereby the slopes of the curves are quite similar.
Results of Bulk Carbon Analyses
Figure 6 summarizes the residual bulk C-contents (measured by combustion analysis/infrared spectroscopy) of all the S304HCu specimens after exposures shown in Figs. 2, 3, and 5. Some additional experiments were carried out using the same temperature/time as described in Figs. 2, 4, however with water vapour contents of 2, 50 or 200 ppm H2O. The C analyses after these exposures are also included in Fig. 6.

Comparison with the data in Figs. 2, 4 and 5 show a clear correlation between the loss in carbon content and the amount of CO detected by the mass spectrometric analyses. The data illustrate that the decarburization increases with increasing temperature. For instance, in the Ar–4%H2 gas with 100 ppm H2O the carbon content in the specimen after decarburization at 1000 °C is around 0.07 wt.%, i.e., hardly different from that in the as received condition. After decarburization at 1050 °C it is around 0.02 wt.% whereas after decarburization at 1100 °C the value is about 0.002 wt.%.
It is easy to estimate that a complete removal of the carbon in the specimen corresponds to a mass loss of approximately 0.3 mg/cm2. This apparently occurs after approximately 12 h total exposure time at 1100 °C; whereas, carbon removal at 1050 °C appears to stop at about 0.15 mg/cm2 (Fig. 5) even though the specimen still contains a certain amount of carbon. Knowing that the specimen contains about 0.02% C (Fig. 6) after decarburization at 1050 °C, the pCO* [Eq. (3)] value at which reaction (2) stops can be estimated from the data in Table 4 calculated for the initial amount of carbon in the steel, i.e., 0.073%. A similar estimate can be made for a specimen decarburized at 1100 °C, which contains 0.0021% C after decarburization. Estimating both pCO* values, one concludes that the rate of the destruction reaction (2) at about pCO* = 2 × 10–4 bar (compared to 7.5 x 10–3 for the original alloy C content) becomes smaller than the rate of oxide formation. So, the specimen starts to gain weight due to oxide formation in spite of the fact that, different from the 1100 °C sample, the carbon in the sample has not yet completely been removed.
The data in Fig. 6 also show, that decarburization increases (more C is removed) with decreasing water vapour content and presence of hydrogen in the atmosphere.
Effect of Specimen Thickness
Decarburization according to reactions (2) and (5) obviously results in depletion of C from the material The time to reach complete depletion of C should be shorter for thin walled steel sections compared to thicker components because of a smaller C-reservoir in the former case. In order to verify this assumption decarburization experiments were performed with specimens of various thicknesses.
Figure 7 shows weight changes as function of time for 1 and 3 mm thick specimens during pre-oxidation and decarburization in the same conditions (pre-oxidation 2 h at 700 °C, decarburization 20 h at 1100 °C in Ar–4%H2–100 ppm H2O). The weight changes for these two specimens are virtually identical up to a total exposure time of approximately 13 h. Subsequently the weight of the 3 mm thick specimen further decreases indicating further decarburization; whereas, the 1 mm specimen starts to exhibit a slight weight gain. This occurs at a total weight loss of approximately 0.3 mg/cm2 which should, based on the considerations above, correspond to a virtually complete decarburization.

a Weight changes and temperature as function of time, and b CO intensities and temperature versus time measured by mass spectrometry during exposure for S304 HCu specimens during 2 h pre-oxidation at 700 °C and subsequent 20 h decarburization at 1100 °C in Ar–4%H2–100 ppm H2O (specimen thickness 1 and 3 mm). Weight change rates during different stages of exposure are inserted in (a)
This is in qualitative agreement with the CO analyses in Fig. 7b and confirmed by the bulk carbon content analyzed by infrared analysis for both specimens after exposure. For the 1 mm thick specimen, a carbon content of 0.002 wt% was measured; whereas, the remaining C content for the 3 mm thick specimen was still 0.04 wt% C (compared to a value in the as-received material of 0.07 wt%).
When looking at the diagram in Fig. 7, it is important to realize that the decarburization reaction already starts at temperatures slightly lower than the final decarburization temperature, in this case 1050 °C. This can be clearly seen by zooming in on the area of heating and the first decarburization hours (Fig. 8).

Enlarged area of pre-oxidation, heating and initial decarburization hours from Fig. 7 (specimen thickness 1 and 3 mm)
Figure 7a shows that for the 1 mm specimen no further weight decrease is observed after a total exposure time of approximately 13 h. In fact, even a slight weight increase occurs, expectedly due to ongoing oxide formation. Depending on the actual decarburization reaction (2 or 4) a slower or faster decarburization rate can be observed. The calculated decarburization rates are presented in Fig. 7a.
Figure 9 shows macro-photographs of the 1 and 3 mm thick specimens. The 1 mm thick specimen is covered by an oxide scale; whereas, no surface oxide was observed on the 3 mm thick specimen. This can be explained in the following way:

Macro-photo image of 1 and 3 mm thick specimens of S304 HCu after decarburization test; 2 h pre-oxidation at 700 °C and subsequently 20 h at 1100 °C in Ar–4%H2–100 ppm (see Fig. 7)
During the pre-oxidation and subsequent heating an oxide scale is formed due to the reaction of the steel surface with the 100 ppm water vapour in the gas. Based on the results presented above, a decarburization starts to occur above approximately 1000 °C, mainly due to a reaction of carbon with the oxide [Eq. (2)]. Apparently, the rate of this reaction is faster than the rate of oxide formation with the effect that the weight gain changes into a weight loss. The weight loss rate is initially around 0.12 mg/cm2 h until the oxide is completely removed by the reaction with carbon. The weight loss rate subsequently decreases to approximately 0.03 mg/cm2 h which indicates that further decarburization is caused by direct reaction of C with water vapour. This reaction continues as long as the carbon content at the steel surface is sufficiently high for Eq. (2) to proceed. After the C is virtually totally removed, the surface starts to oxidize due to reaction with water vapour. For the 1 mm specimen this is the case after a total exposure time of approximately 13 h. The surface oxidation does not occur for the 3 mm specimen up to the maximum exposure time as the remaining C content is still sufficiently high to assure “oxide destruction” by reaction (2).
In both specimens after an initial peak, a lower, slightly decreasing CO signal is observed. The initially fast CO release rate is related to decarburization according to reaction (2), i.e., CO formation due to reduction of chromia and the slower CO release rate after about 7 h total exposure time to the direct reaction of C with water, reaction (5). For the 1 mm thick specimen the CO release decreases to “zero” after approximately 17 h total exposure time.
At the end of exposure, the macroscopic appearance of the 3 mm specimen is metallic bright (Fig. 9) meaning that no apparent oxide formation on the specimen surface occurred at the end of the TG-experiment. This observation confirms that reaction (5) prevails in the 3 mm sample at the end of the test.
For the 1 mm specimen the surface is covered with oxide at the end of the test (Fig. 9). This agrees with no CO release from the specimen and a corresponding slight mass increase after about 17 h (Fig. 7a). Apparently, the CO release becomes virtually “zero” as pCO* becomes too small for oxide removal and surface oxidation prevails. Calculation of the theoretical carbon reservoir in the 20 × 10 × 1 mm thick specimen reveals a value of 0.25 mg/cm2. This value agrees well with the mass change in Fig. 7a (minimum weight change of 0.3 mg/cm2), taking into account that part of the weight gain during heating of the specimen from pre-oxidation to decarburization temperature is related to the buoyancy effect.
Figure 10 shows microstructures of decarburized and as-received specimens after 1000 h exposure at 600 °C in air. In the heat treated as-received material specimen clearly Cr-rich carbide formation mainly at the grain boundaries is observed. This is confirmed by the EDX carbon X-ray mapping presented in Fig. 11, showing clear carbon enrichment at the grain boundaries. For the decarburized specimen only precipitates of NbCrN or Nb-carbonitrides, which are typical for this steel [29], with the size in the range of a few µm are locally present.

Microstructure of decarburized (2 h/700 °C + 20 h/1100 °C, Ar–4%H2-100 ppm H2O) and as-received specimen after 1000 h exposure at 600 °C in air. Optical metallography, specimens etched with NH3 solution. Specimens thickness 1 mm

EDX mappings showing carbon distribution in a decarburized (2 h/700 °C and 20 h/1100 °C, Ar–4%H2–100 ppm H2O) and b as-received specimen after 1000 h at 600 °C
Effect of Pre-oxidation Conditions
Effect of Pre-formed Oxide Thickness
The results presented so far were for specimens continuously exposed to the same atmosphere (Ar–H2–H2O), the pre-oxidation temperature was always 700 °C. In this section, different pre-oxidation temperatures from 700 to 900 °C were used to determine the effect of oxide scale thickness on the decarburization rate. Expecting a thick and dense oxide scale after pre-oxidation (e.g., formed at 900 °C), a higher decarburization temperature such as 1150 °C was also chosen, which, according to thermodynamic predictions, should increase the partial pressure of CO for both decarburization reactions (2) and (5).
Figure 12a shows the pre-oxidation and decarburization for specimens pre-oxidized at various temperatures in Ar–4%H2–100 ppm H2O, the decarburization temperature was 1150 °C. The weight increases at 700 °C after 10 h pre-oxidation is about 0.02 mg/cm2, which (assuming pure chromia formation) corresponds to a scale thickness of 0.12 µm. At 800 °C a very similar result was obtained, while at 900 °C the mass increase was 0.042 mg/cm2, which corresponded to a scale thickness of approximately 0.25 µm. Generally, the shape of the curves for the specimens pre-oxidized at 700 and 800 °C was almost identical both during pre-oxidation and decarburization period (Fig. 12a). The weight change during decarburization started with a steep slope which remained until the end of the experiment. A different behavior was observed for the specimen pre-oxidized at 900 °C. The thicker oxide after the pre-oxidation period slightly retards the onset of decarburization rate (Fig. 12b), while the period of fastest decarburization was longer than for samples pre-oxidized at 700 and 800 °C. Approximately the same slope, i.e., decarburization rate, was observed thereafter for all specimens. The CO analyses presented in Fig. 12b show that the amount of CO produced in case of the 700 and 800 °C specimen is very similar. For the 900 °C specimen the onset of CO formation is shifted in time and the “peak” is much broader than that observed for the two other samples indicating that the onset of C loss is retarded but the overall decarburization is larger for the specimen with the thicker oxide. After the initial “peak”, the CO release rates are virtually identical for all three specimens, in agreement with the TG analyses.

a Weight changes as function of time for S304HCu specimens and b CO intensities versus time measured by mass spectrometer during exposure of S304Hcu specimens; 10 h at various temperatures and subsequently 1h at 1150 °C in Ar–4%H2–100 ppm H2O (Specimens thickness 1 mm). On both diagrams the decarburization area has been enlarged for better visibility
Pre-oxidation in Air
In contrast to the previous investigations, in the next series of experiments the pre-oxidation was done in air, which is in many cases more relevant for practical applications. For this purpose, the samples were exposed in an electrically heated tube furnace for 10 h in laboratory air. Thus, pre-oxidation and subsequent decarburization were separately done (non-continuous exposures). The weight changes after 10 h of pre-oxidation at various temperatures in air are shown in Table 4 revealing the expected higher oxidation rate with increasing temperature. In the following weight change diagrams only the respective isothermal decarburization periods are presented.
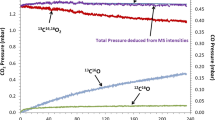
Figure 13 shows the decarburization period for specimens pre-oxidized in air at various temperatures (Table 4). The slight weight loss, which occurred just before the decarburization temperature was reached, was here neglected. Generally, two different trends were observed in the weight change curves depending on pre-oxidation temperature. The specimens pre-oxidized at 700 and 800 °C started with a steep slope in the weight change curve then changing to a less steep, virtually constant slope after the first minutes of exposure. The specimen pre-oxidized at 900 °C shows initially a less steep slope at the beginning of exposure, then changing in the later stage to a much steeper slope. The shape of the weight gain curves for the specimens pre-oxidized at 700 and 800 °C is very similar to that illustrated in Fig. 7 whereby the time until occurrence of the change in slope is slightly longer for the 800 °C specimen. Based on the discussion above, this can be explained by the fact that complete reduction of the oxide scale by reaction with carbon [Eq. (2)] takes a longer time for the thicker 800° C pre-formed scale. After approximately 0.2 h the slopes for both samples become very similar, which would be expected because in both cases it represents the decarburization occurring via direct reaction of the gas with the metal surface. The fundamentally different behavior of the 900 °C pre-oxidized specimen can be explained by the measured evolved CO values shown in Fig. 14. It is important to note that the time axes in Figs. 13 and 14 are different. Figure 13 only shows the decarburization period (1 h), the curve has been corrected for the buoyancy effect (see Sect. ”Experimental”). The graph (Fig. 14) showing the amount of CO escaping from the sample additionally shows both the heating and cooling periods of the specimens, since CO was additionally removed from the specimens during this time. For all studied pre-oxidation temperatures CO formation is detected after the temperature started to rise to 1150 °C. For the specimens pre-oxidized at 700 and 800 °C a rapid CO formation occurs which then declines to lower values (Fig. 14). There is a slight tendency that the amount of CO produced in case of the 800 °C specimen is somewhat larger than that of the 700 °C sample which can be explained by the slight differences in oxide thickness. For the 900 °C specimen the onset of CO formation is shifted in time and the “peak” is much broader than that observed for the two other samples. Generally, the peak detected for specimen pre-oxidized at 900 °C is wider indicating that more CO was removed from the specimens than for the two specimens pre-oxidized at lower temperatures.

SEM images of the surface oxide scale formed on S304HCu during 10 h pre-oxidation at 800 °C in various atmospheres. The images show that a continuous oxide layer of Cr2O3 (fine crystalline areas) is formed in both pre-oxidation atmospheres. On the top of the inner chromia scale spinel crystals are formed

Weight changes as function of time for S304 HCu specimens pre-oxidized for 10 h in air during 1 h decarburization at 1150 °C in Ar–4%H2–100 ppm H2O (specimen thickness 1 mm). Weight change rates in the various stages of exposure are inserted in the figure
This is in qualitative agreement with the proposed mechanism of decarburization. In the used test gas Ar–4%H2–100 ppm H2O the chromia base oxide is thermodynamically stable at 1150 °C and will thus not be reduced by the gas phase during the decarburization step (compare with Table 2). The oxide reduction occurs due to reaction with the carbon in the alloy.
One may argue that the fundamental difference in the time dependence of CO release between the specimens pre-oxidized at 700 or 800 °C on one hand and 900 °C on the other hand may be the result of a fundamentally different oxide morphology. The very low weight gains after 10 h at 700 or 800 °C pre-oxidation might indicate that, contrary to 900 °C, only patches of oxide rather than a continuous, thin oxide layer has been formed. Figure 13 shows that this is not the case. Although thin, the oxides formed during pre-oxidation at 800 °C (Fig. 13) and 700 °C (not shown here) in air and Ar–H2–H2O are continuous and covering the complete surface. The scales formed in the two environments only differ in respect to the type and amounts of spinel crystals on top of the inner chromia scale.
Apparently, the thin oxides formed in air at 700 and 800 °C are sufficiently “defective” or physical defects are rapidly formed by the carbon/chromia reaction so that the formed CO can easily “escape” into the atmosphere resulting in an initial weight loss rate of approximately 0.5 mg/cm2⋅h. Subsequently the decarburization occurs via direct reaction between water vapour and carbon leading to a slower rate of approximately 0.33 mg/cm2⋅h. If a thick, dense oxide is present (such as that formed at 900 °C) the decarburization occurs initially over existing defects in the scale such as microcracks, however apparently the CO cannot easily “escape” resulting in a slow decarburization rate. After some time, the reduction occurring at the steel/oxide interface has created sufficient “damage” in the scale. Thereby the formed CO can easily “escape” resulting in a decarburization rate similar to the initial rate of the two other specimens. The delay time before the onset of rapid decarburization thus logically depends on the properties of the pre-oxidation scale. This mechanism is confirmed by the results shown in Figs. 14 and 15: although the 700 and 800 °C pre-oxidized specimens show initially the highest decarburization rate, the overall carbon loss at the end of the 1150 °C exposure is larger for the 900 °C pre-oxidized sample.

CO intensities versus time measured by mass spectrometry during decarburization of 10 h air pre-oxidized S304 HCu specimens; 1 h at 1150 °C in Ar–4%H2–100 ppm H2O (specimen thickness 1 mm)
Based on the proposed decarburization mechanism of pre-oxidized specimens the weight change and decarburization rates for the various specimens should initially differ. However, after the pre-formed oxide is completely removed, the decarburization rates should be approximately the same for all specimens, independent of the pre-oxidation treatment. This is obviously not yet the case after the 1 h decarburization shown in Fig. 14. Considering, e.g., for the specimen oxidized at 900 °C an oxygen uptake during pre-oxidation of 0,08 mg/cm2 (Table 4), complete reduction of this oxide by reaction (2) reveals a C-loss of 0.059 mg/cm2. The total specimen weight loss for complete reduction of the pre-formed scale (including dissociated Cr2O3 and C depleted from the steel thereby ignoring the additional oxide formation directly after reaching 1150 °C) would be 0.14 mg/cm2. Figure 14 shows that after reaching this value the slope of the weight change curve starts to change toward that of the samples pre-oxidized at 700 and 800 °C.
The specimens pre-oxidized for 10 h at temperatures between 700 and 900 °C in air and subsequently decarburized for 1 h at 1150 °C in Ar–4%H2–100 ppm H2O were analyzed by glow discharge optical emission spectroscopy (GDOES—Horiba Jobin Yvon, HR Profiler), the results are shown in Fig. 16. The diagrams show carbon profiles in weight percent, calculated from the C/Fe ratio compared with the carbon amount in the steel in the as received condition. All profiles show a clear depletion of carbon toward the specimen surface (steep slopes). Additionally, stronger overall carbon depletion is observed for specimens pre-oxidized at higher temperatures which is in agreement with the results presented in Figs. 14 and 15.

GDOES depth profiles of C content as function of depth measured on specimens of S304HCu after 10 h pre-oxidation at 800, 900 and 950 °C respectively in air and subsequently 1 h decarburization at 1150 °C in Ar–4%H2–100 ppm H2O
In order to check if the slope of the weight change rate for the specimen pre-oxidized at 900 °C will after longer time become similar to that of the specimen pre-oxidized at 700 °C (and 800 °C), two additional experiments were carried out. The specimens were again 10 h pre-oxidized in air at 700 and 900 °C, respectively and then decarburized at 1150 °C in Ar–4%H2–100 ppm H2O. The only difference with the previous tests was a longer decarburization time of 2 h. Figure 17 shows excellent reproducibility of the results up to the exposure time of 1 h. It can be seen that after about 1.5 h of decarburization the weight change curves for both specimens start to exhibit a similar slope indicating that after this time the same decarburization reaction/mechanism is responsible for the weight loss.

Weight changes as function of time for pre-oxidized S304 HCu specimens during 1 h (solid lines; data identical to those in Fig. 9) and 2 h (dashed lines) decarburization at 1150 °C in Ar–4%H2–100 ppm H2O (specimen thickness 1 mm). Specimens were pre-oxidized 10 h in air at 700 and 900 °C, respectively
Comparison of Decarburization Rates with and Without Pre-oxidation
Figure 18 compares the decarburization behavior of specimens with and without pre-oxidation at 700 °C in air and in Ar–4%H2–100 ppm H2O. For both pre-oxidized specimens, a rapid weight loss at the beginning of the decarburization exposure is observed. After this first period the two specimens show a smaller weight loss rate which is similar to that of the non-pre-oxidized specimen.

Weight changes as function of time for S304 HCu specimens during 1 h decarburization at 1150 °C in Ar–4%H2–100 ppm H2O (specimen thickness 1 mm). Comparison of specimens without and with 10 h pre-oxidation at 700 °C in H2/H2O and in air respectively
It should be considered that the weight loss for pre-oxidized specimens during decarburization treatment presents the sum of carbon and oxygen (oxide) removed from the specimen. From the decarburization reaction (2) (see theory part) it can be calculated how much C is removed from the specimens using oxygen tied-up in the oxide formed during pre-oxidation. After the pre-oxidation the weight change (oxygen uptake) at 700 °C of the specimen pre-oxidized in air was 0.013 mg/cm2 and of the specimen pre-oxidized in Ar–4%H2–100 ppm H2O 0.014 mg/cm2. Using now the decarburization reaction (2) it can be calculated that for the specimen pre-oxidized (both in air and Ar/H2/H2O) about 0.009 mg/cm2 carbon will be removed from the specimen if the pre-formed oxide is completely removed. Figure 19 shows the carbon loss from the specimen without pre-oxidation and from the specimen after pre-oxidation in air as a function of time. During the initial decarburization period up to about 0.035 h, decarburization in the specimen without oxide follows reaction (5), and in the specimen with pre-formed oxide follows reaction (2). Once the Cr oxide has been completely consumed [Eq. (2)], decarburization will proceed in both samples according to reaction (5), hence the same slope of the curve can be observed after about 0.035 h (Fig. 18). It can be seen that in the case of the pre-oxidized specimen, decarburization proceeds much faster and, at the same time, more carbon is removed from the specimen than in the case of the specimen without the pre-formed oxide.

Carbon loss (calculated from weight change data) as function of time for S304 HCu specimens during initial period of decarburization at 1150 °C in Ar–4%H2–100 ppm H2O (specimen thickness 1 mm). Comparison of specimens without and with 10 h pre-oxidation at 700 °C in H2/H2O and in air respectively, see Fig. 18
Effect of Hydrogen in the Test Atmosphere
From the above results it is apparent that in the case of an existing oxide scale, formed during heating and/or pre-oxidation, rapid decarburization mainly occurs via reaction (2), either directly upon reaching the decarburization temperature or after some delay (see e.g., Figure 15). This would indicate that presence of hydrogen in the atmosphere should not substantially affect the decarburization process. This is, however in contradiction to the results presented in Figs. 2 and 3.
In their studies related to HTR helium Brenner and Graham [25] indicated that it is unlikely that reaction (2) indeed occurs as indicated by the equation, i.e., a solid–solid reaction. They proposed occurrence of a so-called “microclimate” reaction involving reaction of H2 with chromia thereby forming water vapour, the latter reacting with carbon or carbide. It is, however, not easy to understand how this would occur at a free metal surface.
Explaining the results in the present studies concerning the effect of pre-formed oxide thickness on the decarburization, the effect of atmosphere composition, especially the presence of hydrogen, might be explained by a mechanism similar to that proposed by Rahmel/Tobolski [30] as well as Fuji/Meussner [31] for explaining growth rate variations of iron and chromium-based oxide scales in dry and wet gases.
Consider an existing chromia scale on the surface of the austenitic steel S304HCU from which it is known that it always contains some microvoidage (e.g., caused by vacancy condensation), as schematically illustrated in Fig. 20. Growth of the chromia scale mainly occurs via transport of point defects (vacancies and/or interstitials) [32]. In the dry gas reaction (air or Ar–O2), the only transported vapour species within the microvoid in the chromium oxide is O2(g). It is estimated from thermodynamic data that pO2 equals approximately 10–20 bar at 1000 °C (see Table 2). At such a low partial pressure, the transport of O2 in the gap estimated from the Hertz-Langmuir equations [33, 34] is extremely slow and the decarburization reaction (13) is virtually non-existent.

Schematic illustration showing transport of oxygen (a) and hydrogen (b) molecules through the scale and transfer across in-scale void to the specimen surface
In Ar–H2–H2O gases H2(g) molecules can penetrate the scale [34,35,36] react with Cr2O3 and form H2O in the void according to reaction (14).
Again from Table 2 it can be derived that the water vapour content in the void will be in the microbar range, the exact partial pressure depending on the hydrogen partial pressure within the void. According to the Hertz–Langmuir equation [33, 34, 36] the inward H2O transport (Fig. 20b) in the gap is orders of magnitude faster than in the case of O2 during the dry gas reaction. Thus, decarburization by the reaction of water vapour [reaction (5)] will be much faster than that by reaction (13). Thus, the decarburization process occurs by the subsequent reactions (14) and (5) whereby the sum of these two reactions can be represented by reaction (2). Considering that in this context, the Ar–H2O may be an intermediate case between Ar–O2 (or air) and Ar–H2–H2O, the results in Figs. 2 and 3 can at least qualitatively be explained.
Concluding Remarks
In the present work high-temperature decarburization of the austenitic stainless steel S304HCu in Ar–4%H2–xH2O and Ar-xH2O was studied. Emphasis was put on the identification of parameters, which affect the rate of decarburization during heat treatment in Ar–(H2)–H2O-based gas mixtures, thereby addressing a test gas in which the partial pressure of H2 (and H2O) was much larger than that used in previously published investigations related to gas cooled reactor environments.
Thermodynamic considerations revealed that decarburization is possible in low oxygen activity H2/H2O-based atmospheres and is promoted at high temperatures (at and above 1000 °C). Thereby two possible reactions providing sufficiently high CO pressures for effective decarburization were identified. They include direct oxidation of carbon by water vapour at the metal surface as well as reduction of a pre-formed Cr2O3 scale by carbon.
Thermogravimetric experiments were performed with simultaneous measurements of CO release from the specimen using a mass spectrometer. Variation of the test parameters included temperature, specimen thickness as well as atmosphere composition during pre-oxidation and decarburization.
Generally, the rate of decarburization is faster when it occurs by the reaction of Cr2O3 with C from the steel compared to the reaction of C at the steel surface with H2O, provided that in the former case CO can easily evaporate. If during pre-oxidation a thick Cr2O3-scale is formed, the initial CO release is initially found to be slow, indicating a hindered outward CO transport. After some time, however, the pre-formed Cr2O3 scale thickness is reduced and the scale is locally damaged by the reaction with carbon. This enables the evaporation of CO and the total rate of decarburization increases. After even longer exposure time, the pre-formed Cr2O3 is completely consumed and further decarburization occurs via direct reaction of C with the atmosphere (H2O) at a slower rate. This means that selective pre-oxidation may help to minimize decarburization in short-term annealing, however not during long-term heat treatments.
With respect to the effect of the test atmosphere composition, the H2/H2O gas mixtures with low water vapour content (2–10 ppm) induced a more extensive decarburization of pre-oxidized steel specimens compared to the gases with a higher water vapour content (e.g., 100 ppm). During the high-temperature exposure (e.g., at 1000–1100 °C) in Ar–H2–H2O a competition between the oxide formation by water vapour and the oxide reduction by carbon occurs. High water vapour levels will increase the rate of oxide formation and/or the healing of damage caused by the oxide reduction with the result, that above a certain water vapour level decarburization does not occur. Looking at the above results (e.g., Figure 6), it can be said that at 1000 °C adding about 100 ppm water to Ar–4%H2 is sufficient to suppress the decarburization. At 1050 °C, many hundreds of ppm water are required; but in Ar–H2O gas, about 300 ppm is sufficient. At higher temperatures, very high values, probably in the mbar range, would be necessary to significantly reduce the decarburization process.
Increasing decarburization temperature between 1000 and 1150 °C was found to increase the decarburization rate. This is related to an increase in equilibrium partial pressure of CO with increasing temperature for both decarburization reactions (with Cr2O3 and H2O respectively) as revealed by the thermodynamic considerations shown in “Theoretical Background” section.
Testing of specimens with different thickness revealed that in thinner samples with a smaller C-reservoir the complete decarburization occurs earlier than in thick specimens. However, initially, the decarburization rate is practically independent of sample thickness, i.e., kinetics is not determined by C-diffusion in the material but by kinetics of the reactions (2) and (5). After complete decarburization chromia formation on the specimen surface was observed in the test gases with sufficiently high oxygen activities.
It is concluded that the addition of H2 to a water-containing gas significantly accelerates decarburization. This can be explained by the fact that the H2(g) molecules easily penetrate the chromium oxide scale, enter into existing microvoids and thus provide rapid transport of H2O to the specimen surface thereby enhancing decarburization.
References
H.-J. Grabke, E. Marie Müller, and G. Konczos, Scripta Metallurgica 14, 159 (1980).
H. D. Alvarenga, T. V. De Putte, N. Van Steenberge, J. Sietsma, and H. Terryn, Metallurgical and Materials Transactions A 46, 123 (2015).
E.J. Hilinski and G.H. Johnston, in 2014 4th International Electric Drives Production Conference (EDPC), (2014), p. 1.
W. Zeng, Y. Guo, F. Dai, L. Pan, Y. Lei, and T. Liu, Corrosion Science 221, 111251 (2023).
S. Choi and S. V. D. Zwaag, ISIJ International 52, 549 (2012).
N. Birks, G. H. Meier, and F. S. Pettit, Introduction to the High Temperature Oxidation of Metals, (Cambridge University Press, Cambridge, 2006).
K. Sachs and C. W. Tuck, Surface oxidation of steel in industrial furnaces, (ISI Publication 111, The Iron and Steel Institute, London, 1968), pp. 1–17.
W. E. Boggs and R. H. Kachik, Journal of The Electrochemical Society 116, 424 (1969).
M. P. Tonkovič and M. Knez, Materials and Geoenvironment 64, 199 (2017).
R. Shvab, E. Dudrová, O. Bergman, and S. Bengtsson, Powder Metallurgy Progress, 13 (2013).
N. Piperis, Vacuum 22, 387 (1972).
W.J. Quadakkers and H. Schuster, Materials and Corrosion, 36, 141 (1985).
W. J. Quadakkers, Materials and Corrosion 36, 335 (1985).
C. Cabet and F. Rouillard, Journal of Nuclear Materials 392, 235 (2009).
C. Cabet, J. Chapovaloff, F. Rouillard, G. Girardin, D. Kaczorowski, K. Wolski, and M. Pijolat, Journal of Nuclear Materials 375, 173 (2008).
R. R. Adharapurapu, D. Kumar, J. Zhu, C. J. Torbet, G. S. Was, and T. M. Pollock, Metallurgical and Materials Transactions A 42, 1229 (2011).
D. Röhnert, F. Phillipp, H. Reuther, T. Weber, E. Wessel, and M. Schütze, Oxidation of Metals 68, 271 (2007).
W.J. Quadakkers and J. Zurek, in Shreir’s Corrosion, ed. T. Richardson, Vol. 1, (Elsevier, Oxford, 2010).
F. Rouillard, C. Cabet, K. Wolski, and M. Pijolat, Oxidation of Metals 68, 133 (2007).
H. JürgenGrabke and G. Tauber, Archiv für das Eisenhüttenwesen 46, 215 (1975).
K. Bongartz, W. J. Quadakkers, R. Schulten, and H. Nickel, Metallurgical Transactions A 20, 1021 (1989).
C. Wells, W. Batz, and R. F. Mehl, Transactions AIME, Journal of Metals 188, 553 (1950).
H. K. Birnbaum and C. A. Wert, Berichte der Bunsengesellschaft für physikalische Chemie 76, 806 (1972).
W. J. Quadakkers, Materials Science and Engineering 87, 107 (1987).
K. G. E. Brenner and L. W. Graham, Nuclear Technology 66, 404 (1984).
N. Birks and W. Jackson, Journal of The lron and Steel Institute 208, 81 (1970).
W. W. Webb, J. T. Norton, and C. Wagner, Journal of the Electrochemical Society 103, 112 (1956).
M. Michalik, M. Hänsel, and W.J. Quadakkers, in Report Forschungszentrum Jülich, Vol. 67 (2007).
Y. Sawaragi, K. Ogawa, S. Kato, A. Natori, and S. Hirano, The Sumitomo search 48, 50 (1992).
A. Rahmel and J. Tobolski, Corrosion Science 5, 333 (1965).
C. T. Fujii and R. A. Meussner, Journal of the Electrochemical Society 111, 1215 (1964).
D.J. Young, in Corrosion Series, ed. T. Burstein, Vol. 1 (Elsevier Science, Amsterdam, 2008).
P. Kofstad, High Temperature Corrosion, (Elsevier Applied Science, London, 1988).
J. Ehlers, D. J. Young, E. J. Smaardijk, A. K. Tyagi, H. J. Penkalla, L. Singheiser, and W. J. Quadakkers, Corrosion Science 48, 3428 (2006).
J. Żurek, D. J. Young, E. Essuman, M. Hänsel, H. J. Penkalla, L. Niewolak, and W. J. Quadakkers, Materials Science and Engineering A 477, 259 (2008).
I. Langmuir, Physical Review 5, 329 (1913).
Funding
Open Access funding enabled and organized by Projekt DEAL. This study wasn’t funded by any institution.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. All experiments were performed by HC. Material preparation, data collection was performed by JZ. The first draft of the manuscript was written by JZ. WJQ and DN commended the previous version of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zurek, J., Cosler, H., Quadakkers, W.J. et al. Effect of Surface Oxide on Decarburization Reaction in the Austenitic Steel S304HCu. High Temperature Corrosion of mater. 100, 491–523 (2023). https://doi.org/10.1007/s11085-023-10196-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11085-023-10196-w