
Abstract
This study concerns the importance of the precursor (or pre-reactive) state of elementary physicochemical processes whose basic features, as structure, stability, and trapping effect of reagents, are controlled by the balance of intermolecular forces that arise at long range and operate at intermediate and short separation distances. The detailed formulation of such forces, determining formation probability and dynamical evolution of the precursor state, is of relevance in molecular science and difficult to be treated by quantum chemistry. Such a problem has been tackled by us exploiting the phenomenological approach, which employs semi-empirical and empirical formulas to represent strength, range and angular dependence of the leading interaction components involved. In addition to the study of transport phenomena, part of the attention is addressed to chemi-ionization (or Penning ionization) reactions for which neutral reagents lead to atomic and/or molecular ions plus electrons as products. Chemi-ionizations are bimolecular processes occurring in several environments of interest, where a reagent is a species, formed in excited-metastable electronic states by collisions with energetic electrons or cosmic rays. For such reactions all crucial electronic rearrangements, affecting stability and evolution of the weakly bound precursor state, here coincident with the reaction transition state, are characterized with a high detail. The results of the present study are of interest for many other processes, whose precursor states and their relevant features are difficult to characterize, often masked by several other effects.
Graphical abstract

Similar content being viewed by others

1 Introduction
The detailed investigation of strength, range, and nature of the leading components of intermolecular forces is, on general ground, of great relevance in modern research, since these forces control static and dynamical properties of matter, including the role of weakly bound precursor (pre-reactive) states on the evolution of elementary processes (Special Issue 2022a; b; James et al. 2022; Hong et al. 2021a, b; Ascenzi et al. 2019; Falcinelli et al. 2023a). In particular, depending on the stability of adducts controlled by the selectivity of long-range intermolecular forces, the precursor state of barrier-less reactions, such as chemi-ionizations discussed below, coincides with reaction transition states (TS), while in other cases it opens the passage either to the TS of reactive channels with the lowest barrier or to those of less favorable channels.
We have characterized the leading interaction components, operative in several prototypical systems, determining the behavior of the intermolecular potential at long and intermediate separation distance, by performing scattering experiments with the molecular beam technique. The analysis of the experimental observables represents the starting point of our phenomenological approach [denoted phenomenological method in Falcinelli et al. (2023a)], which employs semi-empirical and empirical formulas to represent the Force Fields (FF) in analytical form (Pirani et al. 2006; Capitelli et al. 2007). This possibility, provided for the full range of relative configurations of the interacting partners, is crucial to sharply decrease running times of molecular dynamics simulations of elementary processes, covering from cold up to hot temperature conditions: examples are chemical reactivity and energy transfer in gas phase, either at the gas–solid and at gas–liquid inter-phases (Falcinelli et al. 2023a; Gisler and Nesbitt 2012). In the last years, extensive use has been made of the phenomenological approach to describe static and dynamical properties of systems of increasing complexity and of applied interest (Falcinelli et al. 2023a, and references therein).
Perturbation theory (Maitland et al. 1987) suggests how to partition, at sufficiently large separation distances, the intermolecular potential energy and how to identify the leading components. In particular, we can identify a covalent (or molecular) component, directly related to chemical contributions associated to electronic angular momentum coupling/decoupling and electron/charge transfer effects, and a non-covalent (or supramolecular) component (Weinhold 2023), arising from the combination of induction, electrostatic and van der Waals contributions, where the latter originate from the sum of exchange (or size) repulsion and dispersion attraction. The blend of semi-empirical and empirical formulas provides the dependence of strength and range of the leading components from the fundamental physical properties of the interacting partners (Cambi et al. 1991; Cappelletti et al. 1991; Pirani et al. 2000). Such dependence often favors the discovery of universal scaling laws (Pirani et al. 2006; Capitelli et al. 2007), which are fully exploited by the phenomenological approach. In several cases, its predictions have been tested on and combined with the results of state-of-the-art ab initio calculations, often permitting improvements and further generalizations (Alagia et al. 2011a, b; Cappelletti et al. 2012; Pirani et al. 2019; Nunzi et al. 2020). The phenomenological approach has been recently applied (see also below) to define fundamental features of the precursor or pre-reactive state of important elementary physicochemical processes (Falcinelli et al. 2023a).
In this work, our attention focuses on the role of the precursor state controlling the dynamics of elementary processes, such as the transport phenomena, operative in plasmas, in the planetary atmospheres, and in the interstellar medium. In these environments, chemi-ionization reactions (CHEMI) are basic bimolecular processes playing an important role. CHEMI processes, discovered several years ago (Penning 1927), continue to be of relevance in fundamental and applied research: they provide positive ionic species plus electrons as products and can be considered the inverse of electron attachment processes. Such reactions are often triggered by a reagent excited in high energetic states, formed in collisions with cosmic rays or electrons. Important reagents are noble gas atoms excited in metastable states: at large separation distances from the other partner their behavior is similar to that of alkaline atoms, while at short separation distances the features of the electronic configuration of its ionic core, which is often the same of high electron affinity open shell atoms as halogens, become prominent determining a transition in their chemical behavior. Moreover, when CHEMI is described by optical potentials controlling both the trapping of neutral reagents and the passage from higher neutral channels to lower ionic ones, metastable precursors act as transition states of barrier-less reactions.
Penning ionization and electron attachment processes are relevant in plasmas under several conditions and also of great interest for nuclear fusion. The dissociative attachment to H2, involving the formation of the unstable intermediate molecular ion \({\text{H}}_{2}^{ - }\) in the ground and excited/Rydberg states through Feshbach and shape resonances (Celiberto et al. 2009, 2013, 2023; Capitelli et al. 2006), represents the key process in the mechanism of H− yield in volume negative ion sources, devices designed for the neutral beam injection in fusion reactors (Bacal 2006). Excited states are recognized to play a role in the efficiency of the mechanism: the hypothesis still to be fully demonstrated, to explain the higher H– density observed in experiments consists in the involvement in dissociative attachment of highly excited Rydberg states of H2 (Hassouni et al. 1998; Capitelli et al. 2006). It is also worth mentioning the dissociative recombination process of \({\text{CH}}_{y }^{ + }\) hydrocarbon ions (Chakrabarti et al. 2022), considerably investigated in the literature for their role in fusion plasmas.
The next section emphasizes some basic aspects of the phenomenological approach and the following ones summarize its recent applications, also providing indications for future developments.
2 Foundations of the phenomenological approach
The starting point of our investigation has been the experiments. In particular, projectile-target scattering experiments have been performed with two different apparatuses, exploiting the molecular beam (MB) technique (Pirani et al. 2006, 2008), built in the laboratory started by the late professor G. G. Volpi, to whom this paper is dedicated. Integral and differential cross sections, measured as a function of collision velocity and scattering angle, respectively, show modulations of observables, arising from quantum interference effects; they have been resolved by adopting proper resolution conditions both in scattering angle and in relative collision velocity. Such effects are due to the wave behavior of colliding partners, involved in two-body events, where the intermolecular interaction, originates interference phenomena analogous to those of the light crossing obstacles (Maitland et al. 1987). Frequency and amplitude of oscillating quantum effects are directly related to range, strength and anisotropy of weak intermolecular forces operative at intermediate and large intermolecular distances (Pirani et al. 2006, and references therein).
An Improved Lennard Jones (ILJ) function, useful for the representation of weak interactions under a variety of conditions (Pirani et al. 2004, 2008; Albertí et al. 2005), has been suggested by the analysis of the experimental findings, obtained for several prototype systems. ILJ employs only three basic parameters:
The equilibrium distance rm (or bond length) of the interacting system, the well depth ɛ (or bond energy) and β, related to the partners softness, that defines the shape of the potential well and its angular dependence (Hong et al. 2021b). The ILJ formulation removes most of the inadequacies of venerable LJ potential model, as an excessive short-range repulsion and a too strong long-range attraction. In addition, it provides first and second derivatives of the potential energy in analytical form and this represents a crucial condition to carry out efficient molecular dynamics simulations.
The internally consistent investigation of several prototype systems suggested important relations representing the dependence of attraction and repulsion strength, of ɛ, \(r_{m}\) and β values on fundamental physical properties of interacting partners (Cambi et al 1991; Cappelletti et al. 1991; Capitelli et al. 2007). Such relations represent scaling laws of general validity (Pirani et al. 2006; Capitelli et al. 2007; Nunzi et al. 2020), useful to predict basic interaction features in unknown systems.
The phenomenological approach (Pirani et al. 2006; Capitelli et al. 2007; Falcinelli et al. 2023a) is founded on the analysis of experimental data, combined with the ILJ formulation of the potential energy and with the adoption of scaling laws for the evaluation of range and strength of the leading interaction components. Such method is important since it predicts the behavior of systems of increasing complexity, it suggests new experiments and orients theoretical calculations for testing the method itself, also, it gives insight into the nature of the interaction and to improve and extend the formulation of Force Fields.
3 Transport phenomena
The predictive character of the phenomenological approach has been fully exploited for the calculation of transport cross sections, crucial quantities for evaluating transport coefficients in chemically complex plasmas (Capitelli et al. 2007; Laricchiuta et al. 2007, 2009a, b, 2019; Bruno et al. 2010; Colonna et al. 2018), with impact in the fields of thermal plasmas and aerothermochemistry. For many different technological applications of plasmas, from switches to plasma torches, the reliable modeling of devices relies on the accurate thermodynamic and transport characterization of the plasma, as well as in the fluid dynamic simulation of the heat load at the surface of space vehicles impacting planetary atmospheres during the (re)entry maneuvers. Here, the shock formed at the vehicle nose, with the abrupt change in temperature and pressure conditions, rapidly evolves towards the formation of a complex plasma through the promotion of chemical reactions. The plasma environment is characterized by the extreme complexity of the chemical network, including unstable transient species with high reactivity, e.g., radicals, negative ions and excited states. These chemical species, though a minority, can significantly affect or even drive the plasma evolution; this implies the need to describe, in the framework of the Chapman–Enskog approach for transport properties, the collisional behavior in elastic binary encounters in terms of reduced collision integrals, \(\sigma^{2} {\Omega }^{{\left( {\ell,s} \right)*}}\). The latter result from the thermal averaging of the different components of elastic cross sections, momentum transfer Q(1)(E), viscosity Q(2)(E) and so on. Independently from the adopted scattering approach (classical or quantum based on phase-shifts), the fundamental information relies on the interaction potential and the phenomenological approach provides an accurate, direct and powerful method for the generation of the potential, based on the knowledge of the physical properties of the collisional partners: it allows the calculation of collision integrals with reasonable accuracy for unknown or poorly investigated systems. The method has undeniable advantages: (1) being a full-range model allows the description of the short-range e long-range interaction affecting the collision integral behavior in the high- and low-temperature regions respectively; (2) it reproduces with high accuracy the features of the average potential in the case of open-shell interactions, offering an attractive alternative to the laborious traditional multipotential approach, associated to the large number of molecular states which arise in the interaction of the partners, predictable with the Wigner–Witmer rules of angular momentum composition; (3) the method is apt to the treatment of polyatomic molecules, representing a spatial (angular) averaging of the potential energy surface and reducing the interaction to a unidimensional function depending only on the coordinate of approach; (4) collision integrals, up to high-orders, are straightforwardly available as analytical functions in a wide range of reduced temperatures with errors within a few percentages (Laricchiuta et al. 2007); (5) the framework given by the phenomenological approach responds to requirements of simplicity of the procedure, accuracy of the results and completeness and consistency of the generated database for all the interactions relevant to the plasma system under consideration. As an example of the exceptionally good performances of the method, the phenomenological collision integrals for the interactions of helium with ground state atoms are displayed in Fig. 1 together with the accurate results obtained by Partridge et al. (2001) with a traditional multi-potential approach.

Diffusion- and viscosity-type collision integrals for helium-carbon and helium–oxygen interactions (solid lines) phenomenological approach (markers) multi-potential approach from Partridge et al. (2001)
Another aspect of the phenomenological approach worthy of note is the flexibility to the description of the physics of the interaction, and this has been the basis of its extension to the derivation of transport cross sections for pairs involving excited states. The assessment of the role of excited states in affecting the transport properties of plasmas and the redesign of the scheme for the transition from the traditional ground-state to a novel state-to-state approach, is an important topic in the literature that is gaining new attention nowadays (Buchowiecki and Szabó 2022; Ding et al. 2023). It has been also explored in pioneering works (Capitelli and Ficocelli 1972; Capitelli and Lamanna 1974; Sourd et al. 2007; Laricchiuta et al. 2008). The difficulties described for the interaction of ground-state species are understandably worsened when excited species are considered, in fact open-shell configurations is the norm and the methods in theoretical quantum chemistry have to describe an increasing number of electronic terms for the highly excited quasi-molecule. This explains how important is to show the practicability of the phenomenological approach also for these systems (Laricchiuta et al. 2009b). The low-lying excited states of oxygen and nitrogen atoms and atomic ions, O(3P,1D,1S) O+(4S,2D,2P) and N(4S,2D,2P) N+(3P,1D,1S), in their symmetric (atom–atom) and ion–parent-atom interactions have been derived using the standard phenomenological approach with appropriate use of the excited state polarizability and the number of effective electrons compatible with the excited configurations, finding results comparing well with those obtained in the multi-potential approach. It is important to stress that in the case of these interactions the odd-\(\ell\)-order collision integrals are significantly affected, at high temperatures, by inelastic excitation exchange (charge exchange for atom–ion interactions) (Laricchiuta et al. 2009b; Capitelli et al. 2013); this contribution to the collision integral that would require a close-coupling calculation can be estimated accurately within the asymptotic approach (Kosarim et al. 2006). In the case of hydrogen H(n)–H(n), whose transport significantly affects the plasma properties (Bruno et al. 2007), the peculiarity of the chemical species, connected to the energy spacing of levels, determines a change in the nature of the interaction. When excited states are involved, such a change depends on the progressive approaching of the collisional partners, passing from the attractive interaction between highly polarizable species in the long range, to a Coulomb core-core repulsion: it required an adjustment in the general formulation of the ILJ potential (Laricchiuta et al. 2009b).
The phenomenological approach has largely impacted the community working on the transport in plasmas, being adopted in almost all of the recent publications devoted to the subject, and its breakthrough nature in the field is testified by a very recent paper by the NASA (Bellas Chatzigeorgis et al. 2022) widely leveraging on the method for the derivation of a database of collision integrals for the characterization of a non-ionized C/O/H/Si plasma. The system includes a large number of small organic compounds that originate from the ablation of carbo-phenolic composites in the thermal protection systems, designed to be used in the next-future mission of solar system exploration. In the NASA paper, the number of effective electrons N, required in the correlation formulas, is estimated using two different criteria, both based on ab-initio electronic structure calculations, i.e., one relying on the energy of the atomic orbitals and the other on the atomic radius. Eventually, in (Bellas Chatzigeorgis et al. 2022) N was determined basing on the radius approach, however, the analysis showed that the empirical formula proposed in the phenomenological approach (Cambi et al. 1991) produced values that lie between those obtained with the energy and radius criteria, therefore, this represents a further validation of the method. Considering the case of C2H4, characterized by one of the largest deviations, for the interaction with Ar atoms the experimental value for the parameter (Cd)exp = 97 ± 12 [eV Å6] reported in (Cambi et al. 1991) is to be compared with the theoretical values from the semiempirical formula (Cambi et al. 1991) and from ab-initio calculations (Bellas Chatzigeorgis et al. 2022), that are 103.45 and 91.73, respectively, thus showing approximately the same absolute deviation from the experiment. Different sets of potential parameters derived either using the semiempirical or the accurate ab initio methods reflect in a discrepancy within 10% in the corresponding collision integrals.
4 The weakly bound precursor state of elementary processes
Charge transfer effects (CT), with strength and selectivity not trivial to characterize, often trigger the chemical reactivity (Falcinelli et al. 2023a). The recent investigation of chemi-ionizations (CHEMI) provides basic information on nature, structure and stability of the precursor state, here coincident with reaction transition state (TS). This information appears to be of general interest for many other processes. As stressed in the introduction, CHEMI often involve as a reagent a noble gas atom, excited in a highly energetic metastable state with an electronic configuration np5(n + 1)s1. At large separation distances from the other reagent the chemistry of such metastable atom shows similarity to that of an alkaline atom, while at intermediate and short distance the behavior of its ionic core np5 is emerging, akin to that of a halogen atom with increased electron affinity. Therefore, the precursor state of such reactions plays a role whose characterization is of interest for many other processes affected by electronic rearrangements and CT operative in the TS (Falcinelli et al. 2020a, b, 2023b, c).
Gas phase studies distinguished between phenomena triggered by resonant CT, as the harpooning promoted by collisions of an alkaline (low ionization potential) atom with a halogenated (high electron affinity) species, and phenomena affected by CT in perturbation limit, as structure and stability of noble gas halides and oxides, which are important systems for the UV lasers development (Pirani et al. 2000, 2006; Falcinelli et al. 2017). A crucial-open question is the planning of experiments probing the CT component in the perturbation limit, since such component is often smeared by theoretical methods on other interaction contributions. We performed experiments with high electron affinity open shell fluorine, chlorine and oxygen atoms scattered by closed shell partners (Pirani et al. 2006), with any collision event driven by an interaction affected by CT.
The apparatus, described in detail in several previous papers (Pirani et al. 2006; Falcinelli et al. 2017, 2023a, and references therein), exploited a microwave discharge source, generating the open shell atomic beam analyzed, along its path, in velocity by a mechanical selector, in intensity by an online mass spectrometer detector. The magnetic probe, performed with a Stern–Gerlach magnetic selector, operating inserted along the beam path, has been important to provide information on the electronic state of generated atoms, on the distribution of their spin–orbit levels, identified by the electronic quantum number J, and also it allowed permitted a controlled variation of the to vary in a controlled way atomic magnetic sublevel populations of the atomic beam. The analysis of collision experiments, carried out introducing a gas target into the scattering chamber inserted along the beam path, provided information on the different stability of collision complexes formed with Σ and Π symmetries. For the cases involving projectile open-shell halogen atoms that approach to an isotropic partner, such different symmetries correspond respectively, to their half-filled orbital aligned parallel and perpendicular to the separation distance R.
In particular, the detailed analysis of experimental findings provided all basic features of VΣ and VΠ interactions. It is found that while VΠ is a pure vdW interaction, while VΣ is stabilized by a chemical component that arises from a configuration interaction between the ground neutral and the excited ionic state of the same symmetry, differing for one electron exchange (Aquilanti et al. 1997; Pirani et al. 2000). Therefore, an appreciable CT contribution stabilizes the system. The experimental findings, obtained under the same conditions for several systems, suggested the CT dependence on the ionization potential and on the electron affinity of the electron donor and of the electron acceptor, respectively. This dependence represents a further scaling law (Pirani et al. 2006; Nunzi et al. 2020; Falcinelli et al. 2023a), of relevance to describe the transition from intermolecular bonds of different nature.
5 The stereodynamics of prototype CHEMI
Information on electronic rearrangements promoted by collisions of open shell atoms is of interest also for CHEMI. As indicated in the upper part of the Fig. 2, CHEMI such reactions are promoted by energetic open shell species (X*), highly excited in a metastable electronic state, colliding with another atomic/molecular reaction partner M. The (X···M)* precursor state shows involves a Rydberg electron with whose a floppy cloud wraps wrapping an ionic core. The latter is described by two limit electronic configurations, X+-M and X-M+, different in energy and coupled by CT. The reaction is triggered by the relaxation of the system in the lowest state. The energy release stimulates the ejection of is sufficient to eject an external electron with a defined kinetic energy. Other products are the associated ion XM+, the parent or penning ion M+ and its fragmentation species (Brunetti and Vecchiocattivi 1993; Siska 1993).

(Partially adapted from Fig. 5 of (Falcinelli et al. 2023a)) Upper part: a representation of CHEMI. Lower and intermediate parts: A Ne*(3P) atom approaches to an Ng partner. The cartoon suggests important details of the direct (intermediate part) and indirect (lower part) mechanisms stimulated by chemical and chemical–physical forces, respectively. The arrows of different colors indicate the different electronic rearrangements accompanying the two mechanisms. For major details see below and (Falcinelli et al. 2023a)
If X* is a metastable helium or neon atom, the reaction occurs with all molecules and also with Ar, Kr and Xe, promoting, in the last case, prototype atom–atom CHEMI. They have been the target of several experimental and theoretical studies and on them we addressed particular attention to obtain information of general interest also for more complex processes (Falcinelli et al. 2020a). Figure 2 shows the Ne*(3P) reagent, whose excited electron is in the “floppy” 3s orbital and the ionic core (having electronic configuration 2p5) is a fluorine atom with increased electron affinity, that approaches to another Ng (Noble gas) partner. If the precursor state, formed at low collision energies, assumes the structure of a weak interacting diatom, the perturbed X* tends to break the validity of the optical selection rules. Under such conditions the reaction can be triggered by a quantum of energy associated to a virtual photon exchange between reagents (Miller 1970; Miller and Morgner 1977; Brunetti and Vecchiocattivi 1993; Siska 1993), becoming a photo-ionization process (see lower part of the Fig. 2). Instead, if the precursor state, formed at high collision energies, is a true chemical molecule, the increased overlap between valence orbital of reagents stimulates a direct electron transfer process and CHEMI can be classified (Falcinelli et al. 2020a) as elementary exothermic oxidation reaction (see intermediate part of the Fig. 2).
As nuclear processes, also CHEMI are driven by an optical potential W. In particular, its real component V controls the dynamical approach of reagents, while the imaginary component Γ, or resonance width, defines the “opacity” of the investigated system, since it is triggering the passage from neutral reagents to ionic products, through valence electron rearrangements (Falcinelli et al. 2020a). As stressed above, CHEMI are barrier-less reactions, with TS coincident with the precursor state, of interest in fundamental and applied research. In particular, they are of relevance to properly define the probability of reactivity events at low temperature (Dulieu and Osterwalder 2018); to evaluate the relevance of the quantum nature of matter (Arango et al. 2006); to control the hot chemistry in flames and plasmas; to exploit new soft ionization devices in mass spectrometric detection (Falcinelli et al. 2020a, b, and references therein).
Scattering experiments have been performed in the Perugia Laboratory and the phenomenological approach provided all basic features of the precursor-transition state (Brunetti and Vecchiocattivi 1993; Falcinelli et al. 2020a, b, 2023a, b). The results of the theoretical treatment have been tested on the experimental observables obtained in our and in other laboratories.
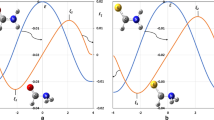
The phenomenological approach suggests that the collisions of reagents generate the precursor state caged within, a sort of electromagnetic trap where the quantum confinement of the system affects all electron rearrangements promoting the reaction (Falcinelli et al. 2021a, 2023a). Such rearrangements depend on the intermolecular forces operative in the trap, stimulating electronic cloud polarization, charge transfer, spin–orbit effects, their perturbations on involved open shell species, and coupling/decoupling of angular momentum arising from electronic components combined with contributions of other nature. Figure 3 emphasizes the electronic polarization for Ne*(3P) approaching to another Ng and suggests that the internal ionic state, formed for collisions of reagents in the entrance channel, and the state of products in the exit channel, are coupled by CT, as in F, Cl-Ng systems. Moreover, colliding reagents generate adducts rotating at separation distance variable. Therefore, angular momentum couplings accompanying atom–atom reactions can be represented as proper sequences of Hund’s cases of rotating diatomic molecules (Falcinelli et al. 2021b).

(Partially adapted from Fig. 6 of (Falcinelli et al. 2023a)) Upper part: the electromagnetic trap controlling precursor state formation of atom–atom CHEMI. Intermediate part: internal ionic states of entrance and exit channels coupled by CT (Aquilanti et al. 1997). Lower part: collision complex, confined in the Hund’s cases (a) and (c), and quantum numbers adopted to define electronic angular momentum couplings. For major information on the transition between different schemes of angular momentum couplings see (Pirani et al. 2006; Falcinelli et al. 2021b)
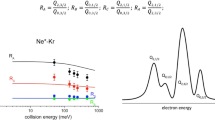
The apparatus, operative in the Perugia laboratory, adopts again the MB technique (Brunetti and Vecchiocattivi 1993). It exploits a noble gas beam source and the emerging atoms, excited by a microwave discharge or by collisions with electrons, cross at right angle the beam of the target gas. A hemispherical electron analyzer, put above the crossing region, is used to measure the Penning Ionization Electron Spectra (PIES), that define intensity and kinetic energy of emitted electrons (Brunetti et al. 2013; Falcinelli et al. 2018). All other ionic products are detected by a mass spectrometer, from whose signals total, partial ionization cross sections and branching ratios, are obtained. For krypton and xenon targets, measured PIES resolved the different collision energy dependence of the probability of reaction channels opened by spin–orbit levels of Ne*(3PJ) reagent and forming Kr+ and Xe+ (2PJ) ionic products in specific J states (Falcinelli et al. 2018). PIES measurements represent then a “sort” of transition state spectroscopy. This finding more clearly emerges when the approach of a Ne* atom to a water molecule at defined collision energy is considered. The PIES, measured at a defined collision energy (see Fig. 4), shows two main peaks related to the formation of water ion in the ground and in the first excited state that occurs through different reaction channels whose precursor-transition states are found to exhibit different energy and structure (Brunetti et al. 2013; Falcinelli et al. 2016).

PIES measured at a defined collision energy (40 meV). A depicture of the different symmetry of the precursor states, opening the reaction channels leading to the formation of water ion in the ground and in the first excited electronic state, is given in the upper part
All valence electron rearrangements promoted by intermolecular forces are accompanied by adiabatic and non adiabatic effects: they control, respectively, collision dynamics and reactivity (Falcinelli et al. 2018, 2020a). The adiabatic effects determine basic features of the real potential in entrance channels and of the interaction in exit channels, where the configuration interaction by CT dominates (Falcinelli et al. 2020b). The adiabatic potentials \(V_{|J,\Omega > }\) account for the conversion of atomic quantum states, represented as |J,Ω˃, with Ω defining the absolute projection J along R, into molecular states of Σ and Π symmetry.
Adiabatic potentials have been obtained in internally consistent way for Ne*-Ar, Ne*-Kr and Ne*-Xe systems. Their comparison, discussed extensively in recent papers (Falcinelli et al. 2020a, 2023a), is useful to emphasize the change in the interaction anisotropy and the different role of the spin–orbit coupling in the exit channels.
Non adiabatic effects, promoting the reactivity through the Γ component, are stimulated by the same electronic rearrangements. Therefore, real and imaginary parts of the optical potential must be interdependent. The phenomenological approach provides two coefficients, linking adiabatic and non adiabatic effects (Falcinelli et al. 2020a, b, 2023a). Such coefficients, labeled as Cx and Cy, quantify, respectively, the Σ character degree in each entrance and each exit channel at each separation distance. They represent marker-tracings of the electronic evolution with its dependence on the separation distance and are the equivalent of indicators in acid–base reactions useful to indicate their pH character. For instance, the Cx behavior, associated to selected entrance channels, suggests that at sufficiently large separation distances, where Cx assumes the asymptotic 1/3 and 2/3 values, the reagents are so separated that intermolecular forces vanish and no reaction occurs. A first critical region is where Cx starts to vary, the electronic rearrangements stimulate the reactions of two atoms perturbed by weak long-range forces. Other two critical regions are where Cx strongly varies and where it assumes 1 or 0, as constant value. Here chemical forces emerge and become dominant. The precursor state passes from a diatomic to a molecular structure, determining a different reaction mechanism.
Accordingly, we identified (Falcinelli et al. 2018, 2020a):
-
A direct mechanism, driven by chemical interactions, whose AΣ–Σ and AΠ–Π nonadiabatic coupling terms depend on the molecular character assumed by initial and final states.
-
An indirect mechanism, whose AΣ–Π and AΠ–Σ non adiabatic coupling terms, basically promoted by polarization, spin–orbit interactions and Coriolis contributions, exhibit a less pronounced radial dependence respect to AΣ–Σ and AΠ–Π. The indirect mechanism dominates at large separation distances, where weak interaction components can stimulate a photo-ionization process.
Therefore, state to state imaginary Γ components are defined as weighted sum of AΛ–Λ′ coupling terms, whose relative weights depend on Cx and Cy coefficients (Falcinelli et al. 2020b).
Internally consistent optical potential formulations have been proposed for Ne*-Ar, Ne*-Kr and Ne*-Xe, allowing a critical comparison between predicted and measured scattering properties. For such reagents, calculated collision energy dependent state-to-state total ionization cross sections emphasize their anisotropy and their dependence on real and imaginary components, operative along each selected channel (Falcinelli et al. 2020a, b). The statistical average over all channels provides cross section results in good agreement with experimental data (Gregor and Siska 1981), measured without state selection. The same analysis gives also state-to-state Associative/Penning ratios, that is the formation probability of an associated ion (as for instance NeAr+) respect to an atomic ion (as Ar+). Each ratio depends on the position of the potential well in exit channel, trapping the formed associated ion, respect to the region of entrance channel where the reaction effectively occurs. Predicted ratios are consistent with state averaged results, measured in Perugia laboratories (Aguilar-Navarro et al. 1985). They also agree with ratios measured in Lausanne by Osterwalder group (Gordon et al. 2017, 2018; Zou et al. 2018), using beams of Ne*(3P2) atoms selected in their |J,Ω˃ quantum states.
Recently, the study focused on CHEMI involving prototype Ne*-diatomic molecules (M) as reagents (Falcinelli et al. 2021b, 2023c). Their description complicates since real and imaginary parts depend also on the M orientation and the associated ion, NeM+, can pre-dissociate if the M+ moiety forms vibration-rotationally excited.
The permanent electric quadrupole moment of N2 determines structures of the precursor state rather different respect to that controlling atom–atom reactions (see Fig. 5).

(Partially adapted from Fig. 13 of (Falcinelli et al. 2023a)) The precursor state of Ne* + N2 CHEMI formed by collisions in the collinear configuration
The phenomenological approach provided real and imaginary parts of the optical potential for Ne* + N2 CHEMI, including their dependence on the molecular orientation and on the atomic sublevel involved. Consistently, the interaction in the exit channels has been obtained and the role of direct and indirect mechanisms has been quantified (Falcinelli et al. 2021c, 2023a, b). The proposed treatment reproduces:
-
total (elastic + inelastic) integral cross sections, measured as a function of the collision velocity in the Eindhoven laboratory with the resolution of a maximum due quantum interference effect in the elastic scattering controlled by the long-range interaction (Kerstel et al. 1988);
-
total ionization cross sections, measured in Perugia and in Eindhoven laboratories and controlled by the optical potential at intermediate and short separation distances (Aguilar et al. 1990; Van den Berg et al. 1987);
-
the larger reactivity, at high collision energy, of 3P0 respect to 3P2, because the faster increase of the Cx marker-tracing associated to the precursor state formed by the 3P0 reagent (Van den Berg et al. 1987).
6 Perspectives and conclusions
The first next target is the extension of the phenomenological approach to the study of more complex processes, starting from CHEMI involving hetero-nuclear diatomic molecule and their comparison with CHEMI of homo-nuclear cases (Zou and Osterwalder 2020). Chemistry and physics of CO is of great interest for its peculiar role since this molecule participates to elementary processes occurring in several environments, as plasmas, flames and planetary atmospheres. For Ne* + CO the construction of the optical potential, both in the real and imaginary components with their dependence on the atomic sublevel and on the molecular orientation, is in progress. Sequence and shape of CO HOMO orbital suggest that, differently from N2, the precursor state at 0° and 180° exhibits pronounced anisotropy that should be reflected in the optical potential.
The second target concerns the full “topology” of the reaction stereo-dynamics, controlled by molecular orientation, centrifugal barrier, and capture effects by long-range attractions. This target is of basic interest not only for CHEMI but also for myriads of other elementary processes (Perreault et al. 2017; Willitsch 2017; Tsikritea et al. 2022). In particular, the rationalization of non-Arrhenius behavior (De Fazio et al. 2019, 2020; Kurnosov et al. 2017) is of crucial relevance for processes occurring at low temperature.
Concerning the use of the phenomenological approach in applications, such as specifically to estimate the collision integrals encountered in calculations of transport properties, the most attractive envisaged extension is the systematic treatment of excited state interactions to allow the full passage to a state-to-state scheme.
Finally, from the general point of view of investigations of elementary physicochemical processes induced by intermolecular forces of weak and intermediate strength, the controlled growth of clusters involving aromatic species is of fundamental interest for synthesis and use of nanostructures (James et al. 2022). Also, clusters of molecules around inorganic ions represent good candidates for storage and recovery of gaseous species such as hydrogen (García-Arroyo et al. 2023).
Data availability
All data generated or analysed during this study are included in this published article and related references.
References
Aguilar A, Brunetti B, Gonzalez M, Vecchiocattivi F (1990) A crossed beam study of the ionization of molecules by metastable neon atoms. Chem Phys 145:211–218
Aguilar-Navarro A, Brunetti B, Rosi S, Vecchiocattivi F, Volpi GG (1985) Velocity dependence of the cross section for Penning and associative ionization of argon atoms by metastable neon atoms. J Chem Phys 82:773–779
Alagia M, Candori P, Falcinelli S, Pirani F, Pedrosa Mundim MS, Richter R, Rosi M, Stranges S, Vecchiocattivi F (2011a) Dissociative double photoionization of benzene molecules in the 26–33 eV energy range. Phys Chem Chem Phys 13:8245
Alagia M, Candori P, Falcinelli S, Mundim MSP, Pirani F, Richter R, Rosi M, Stranges S, Vecchiocattivi F (2011b) Dissociative double photoionization of singly deuterated benzene molecules in the 26–33 eV energy range. J Chem Phys 135:144304
Albertí M, Castro A, Laganà A, Moix M, Pirani F, Cappelletti D, Liuti G (2005) A molecular dynamics investigation of rare-gas solvated cation-benzene clusters using a new potential model. J Phys Chem A 109:2906–2911
Aquilanti V, Cappelletti D, Pirani F (1997) Bond stabilization by charge transfer: the transition from van der Waals forces to the simplest chemical bonds. Chem Phys Lett 271:216–222
Arango CA, Shapiro M, Brumer P (2006) Cold atomic collisions: coherent control of penning and associative ionization. Phys Rev Lett 97:193202
Ascenzi A, Cernuto A, Balucani N, Tosi P, Ceccarelli C, Martini LM, Pirani F (2019) Destruction of dimethyl ether and methyl formate by collisions with He+. A&a 625(A72):1–9
Bacal M (2006) Physics aspects of negative ion sources. Nucl Fusion 46:S250
Bellas Chatzigeorgis G, Haskins JB, Scoggins JB (2022) Transport properties for neutral C, H, N, O, and Si-containing species and mixtures from the Gordon and McBride thermodynamic database. Phys Fluids 34:087106
Brunetti B, Vecchiocattivi F (1993) Autoionization dynamics of collisional complexes. In: Ng C Y, Baer T, Powis I, (eds) Current topics in ion chemistry and physics, Wiley, New York, pp 359–445
Brunetti BG, Candori P, Falcinelli S, Pirani F, Vecchiocattivi F (2013) The stereodynamics of the Penning ionization of water by metastable neon atoms. J Chem Phys 139:164305
Bruno D, Laricchiuta A, Capitelli M, Catalfamo C (2007) Effect of electronic excited states on transport in magnetized hydrogen plasma. Phys Plasmas 14(2):022303
Bruno D, Catalfamo C, Capitelli M, Colonna G, De Pascale O, Diomede P, Gorse C, Laricchiuta A, Longo S, Giordano D, Pirani F (2010) Transport properties of high-temperature Jupiter atmosphere components. Phys Plasmas 17(11):112315
Buchowiecki M, Szabó P (2022) N-H collision integrals with study of repulsive interactions. Plasma Sour Sci Technol 31(4):045010
Cambi R, Cappelletti D, Liuti G, Pirani F (1991) Generalized correlations in terms of polarizability for van der Waals interaction potential parameter calculations. J Chem Phys 95:1852–1861
Capitelli M, Ficocelli V (1972) Collision integrals of oxygen atoms in different electronic states. J Phys B 5(11):2066
Capitelli M, Lamanna U (1974) Collision integrals of electronically excited states and transport coefficients of thermal plasmas. J Plasma Phys 12(1):71–79
Capitelli M et al (2006) Vibrational kinetics, electron dynamics and elementary processes in H2 and D2 plasmas for negative ion production: modelling aspects. Nucl Fusion 46(6):S260
Capitelli M, Cappelletti D, Colonna G, Gorse C, Laricchiuta A, Liuti G, Longo S, Pirani F (2007) On the possibility of using model potentials for collision integral calculations of interest for planetary atmospheres. Chem Phys 338:62–68
Capitelli M, Bruno D, Laricchiuta A (2013) Fundamental aspects of plasma chemical physics: transport, vol 74. Springer Science & Business Media, Berlin
Cappelletti D, Liuti G, Pirani F (1991) Generalization to ion-neutral systems of the polarizability correlations for interaction potential parameters. Chem Phys Lett 183:297–303
Cappelletti D, Ronca E, Belpassi L, Tarantelli F, Pirani F (2012) Revealing charge-transfer effects in gas-phase water chemistry. Acc Chem Res 45:1571–1580
Celiberto R, Janev RK, Wadehra JM, Laricchiuta A (2009) Cross sections for 14-eV e-H2 resonant collisions: dissociative electron attachment. Phys Rev A 80:012712
Celiberto R, Janev RK, Laporta V, Tennyson J, Wadehra JM (2013) Electron-impact vibrational excitation of vibrationally excited H2 molecules involving the resonant \(^{2}\Sigma_{g}^{+}\) Rydberg-excited electronic state. Phys Rev A 88:062701
Celiberto R, Capitelli M, Laricchiuta A, Pietanza LD, Colonna G (2023) Advanced models for negative ion production in hydrogen ion sources. In: Bacal M (ed) Chapter 7 in physics and applications of hydrogen negative ion sources. Springer series on atomic, optical, and plasma physics, vol 124. Springer, Cham
Chakrabarti K, Mezei JZ, Schneider IF, Tennyson J (2022) Electron collision studies on the molecular ion. J Phys B 55(9):095201
Colonna G, D’Angola A, Pietanza LD, Capitelli M, Pirani F, Stevanato E, Laricchiuta A (2018) Thermodynamic and transport properties of plasmas including silicon-based compounds. Plasma Sour Sci Technol 27:015007
De Fazio D, Aquilanti V, Cavalli S (2019) Quantum dynamics and kinetics of the F + H2 and F + D2 reactions at low and ultra-low temperatures. Front Chem 7:328
De Fazio D, Aquilanti V, Cavalli S (2020) Benchmark quantum kinetics at low temperatures toward absolute zero and role of entrance channel wells on tunneling, virtual states, and resonances: the F + HD reaction. J Phys Chem A 124:12–20
Ding Z, Qin Z, Liu L (2023) Collision integrals for N(4S)–N(4S), N(4S)–N(2D), and N(4S)–N(2P) interactions. Phys Fluids 35(2):027127
Dulieu O, Osterwalder A (2018) Cold chemistry molecular scattering and reactivity near absolute zero. Royal Society of Chemistry, Cambridge, pp 1–670
Falcinelli S, Bartocci A, Cavalli S, Pirani F, Vecchiocattivi F (2016) Stereodynamics in the collisional autoionization of water, ammonia, and hydrogen sulfide with metastable rare gas atoms: competition between intermolecular halogen and hydrogen bonds. Chem Eur J 22:764–771
Falcinelli S, Candori P, Pirani F, Vecchiocattivi F (2017) The role of charge transfer in the stability and reactivity of chemical systems from experimental findings. Phys Chem Chem Phys 19:6933–6944
Falcinelli S, Vecchiocattivi F, Pirani F (2018) Adiabatic and nonadiabatic effects in the transition states of state-to-state autoionization processes. Phys Rev Lett 121:163403
Falcinelli S, Farrar JM, Vecchiocattivi F, Pirani F (2020a) Quantum-state controlled reaction channels in chemi-ionization processes: radiative (optical−physical) and exchange (oxidative−chemical) mechanisms. Acc Chem Res 53:2248–2260
Falcinelli S, Vecchiocattivi F, Pirani F (2020b) General treatment for stereo-dynamics of state-to-state chemi-ionization reactions. Commun Chem 3:64
Falcinelli S, Vecchiocattivi F, Cavalli S, Pirani F (2021a) Precursor state of chemi-ionization reactions and confinement of valence electrons by anisotropic intermolecular forces. Eur Phys J D 75:94
Falcinelli S, Vecchiocattivi F, Pirani F (2021b) Electronic rearrangements and angular momentum couplings in quantum state-to-state channels of prototype oxidation processes. J Phys Chem A 125:1461–1467
Falcinelli S, Vecchiocattivi F, Pirani F (2021c) Selectivity of weak intermolecular forces and precursor state of elementary oxidation reactions, a new insight on Ne* + N2chemiionization. Sci Rep 11:19105
Falcinelli S, Cappelletti D, Vecchiocattivi F, Pirani F (2023a) The role of the precursor state on the stereo-dynamics of elementary processes. Phys Chem Chem Phys 25:16176–16200
Falcinelli S, Vecchiocattivi F, Pirani F (2023b) The topology of the reaction stereo-dynamics in chemi-ionizations. Comm Chemistry 6:30
Falcinelli S, Parrani M, Vecchiocattivi F, Pirani F (2023c) The selective role of the orbital angular momentum on the reaction stereo-dynamics. Eur Phys J D 77:65
García-Arroyo E, Campos-Martínez J, Bartolomei M, Hernández MI, Pirani F, Halberstadt N (2023) Attachment of hydrogen molecules to atomic ions (Na+, Cl−): examination of an adiabatic separation of the H2 rotational motion. PhysChemPhys. https://doi.org/10.1002/cphc.202300424
Gisler AW, Nesbitt DJ (2012) On probing ions at gas-liquid interface by quantum state resolved molecular beam scattering: the curious incident of the cation in the night time. Faraday Discuss 157:297–305
Gordon SDS, Zou J, Tanteri S, Jankunas J, Osterwalder A (2017) Energy dependent stereodynamics of the Ne(3P2) +Ar reaction. Phys Rev Lett 119:053001
Gordon SDS, Omiste JJ, Zou J, Tanteri S, Brumer P, Osterwalder A (2018) Quantum-state-controlled channel branching in cold Ne(3P2) +Ar chemi-ionization. Nat Chem 10:1190–1195
Gregor RW, Siska PE (1981) Differential elastic scattering of Ne*(3s3P2,0) by Ar, Kr, and Xe: optical potentials and their orbital interpretation. J Chem Phys 74:1078–1092
Hassouni K, Gicquel A, Capitelli M (1998) The role of dissociative attachment from Rydberg states in enhancing H− concentration in moderate-and low-pressure H2 plasma sources. Chem Phys Lett 290(4–6):502–508
Hong Q, Bartolomei M, Esposito F, Coletti C, Sun Q, Pirani F (2021a) Reconciling experimental and theoretical vibrational deactivation in low-energy O + N2 collisions. Phys Chem Chem Phys 23:15475–15479
Hong Q, Sun Q, Pirani F, Valentín-Rodríguez MA, Hernández-Lamoneda R, Coletti C, Hernández MI, Bartolomei M (2021b) Energy exchange rate coefficients from vibrational inelastic O2\(^{3}\Sigma_{g}^{-}\) + O2\(^{3}\Sigma_{g}^{-}\) collisions on a new spin-averaged potential energy surface. J Chem Phys 154:064304
James A, Jones C, Melekamburath A, Rajeevan M, Srinivasamurthy Swathi R (2022) A journey toward the heaven of chemical fidelity of intermolecular force fields. Wires Comput Mol Sci e1599:1–29
Kerstel ERT, Janssens MFM, van Leuwen KAH, BeijerinckH CW (1988) Intermolecular potentials for the metastable Ne*-rare gas and Ne*-molecule systems. Chem Phys 119(2–3):325–341
Kosarim AV, Smirnov BM, Capitelli M, Celiberto R, Laricchiuta A (2006) Resonant charge exchange involving electronically excited states of nitrogen atoms and ions. Phys Rev A 74(6):062707
Kurnosov A, Cacciatore M, Pirani F, Laganà A, Martí C, Garcia E, (2017) Closer versus long range interaction effects on the non-arrhenius behavior of quasi-resonant O2 + N2 collisions. J Phys Chem A 121:5088–5099
Laricchiuta A, Colonna G, Bruno D, Celiberto R, Gorse C, Pirani F, Capitelli M (2007) Classical transport collision integrals for a Lennard-Jones like phenomenological model potential. Chem Phys Lett 445:133–139
Laricchiuta A, Bruno D, Capitelli M, Celiberto R, Gorse C, Pintus G (2008) Collision integrals of oxygen atoms and ions in electronically excited states. Chem Phys 344(1–2):13–20
Laricchiuta A, Bruno D, Capitelli M, Catalfamo C, Celiberto R, Colonna G, Diomede P, Giordano D, Gorse C, Longo S, Pagano D, Pirani F (2009a) High temperature Mars atmosphere. Part I: transport cross sections. Eur J Phys D 54:607–612
Laricchiuta A, Pirani F, Colonna G, Bruno D, Gorse C, Celiberto R, Capitelli M (2009b) Collision integrals for interactions involving atoms in electronically excited states. J Phys Chem A 113:15250–15256
Laricchiuta A, D’Angola A, Pirani F, Pietanza LD, Capitelli M, Colonna G (2019) Thermodynamic and transport properties of reacting air including ablated species. In: Colonna G, Capitelli M, Laricchiuta A, (eds) Hypersonic meteoroid entry physics, IOP Publishing, Bristol, pp 1–14
Maitland GC, Rigby M, Smith B, Wakeham WA (1987) Intermolecular forces their origin and determination. Clarendon press, Oxford
Miller WH (1970) Theory of Penning ionization. I. Atoms J Chem Phys 52:3563–3572
Miller WH, Morgner H (1977) A unified treatment of Penning ionization and excitation transfer. J Chem Phys 67:4923–4930
Nunzi F, Pannacci G, Tarantelli F, Belpassi L, Cappelletti D, Falcinelli S, Pirani F (2020) Leading interaction components in the structure and reactivity of noble gases compounds. Molecules 25:2367
Partridge H, Stallcop JR, Levin E (2001) Potential energy curves and transport properties for the interaction of He with other ground-state atoms. J Chem Phys 115(14):6471–6488
Penning FM (1927) Über Ionisation durch metastabile Atome. Naturwissenschaften 15:818
Perreault WE, Mukherjee N, Zare RN (2017) Quantum control of molecular collisions at 1 kelvin. Science 358:356–359
Pirani F, Giulivi A, Cappelletti D, Aquilanti V (2000) Coupling by charge transfer: role in bond stabilization for open-shell systems and ionic molecole and in harpooning and proton attachment processes. Mol Phys 98:1749–1762
Pirani F, Albertí M, Castro A, Moix Teixidor M, Cappelletti D (2004) Atom-bond pairwise additive representation for intermolecular potential energy surfaces. Chem Phys Lett 394:37–44
Pirani F, Maciel GS, Cappelletti D, Aquilanti V (2006) Experimental benchmarks and phenomenology of interatomic forces: open shell and electronic anisotropy effects. Int Rev Phys Chem 25:165–199
Pirani F, Brizi S, Roncaratti LF, Casavecchia P, Cappelletti D, Vecchicattivi F (2008) Beyond the Lennard–Jones model: a simple and accurate potential function probed by high resolution scattering data useful for molecular dynamics simulations. Phys Chem Chem Phys 10:5489–5503
Pirani F, Cappelletti D, Falcinelli S, Cesario D, Nunzi F, Belpassi L, Tarantelli F (2019) Selective emergence of the halogen bond in ground and excited states of noble-gas–chlorine systems. Angew Chem Int Ed 58:4195–4199
Siska PE (1993) Molecular-beam studies of Penning ionization. Rev Mod Phys 65:337–412
Sourd B, André P, Aubreton J, Elchinger MF (2007) Influence of the excited states of atomic nitrogen N(2D) and N(2P) on the transport properties of nitrogen. Part i: atomic nitrogen properties. Plasma Chem Plasma Proc 27:35–50
Special Issue (2022) Intermolecular forces: from atoms and molecules to nanostructures. Molecules 27:3072
Special Issue (2022a) Long-range intermolecular interactions in chemistry and physics. Chem Phys Lett 32:96
Tsikritea A, Diprose JA, Softley TP, Heazlewood BR (2022) Capture theory models: an overview of their development, experimental verification, and applications to ion-molecule reactions. J Chem Phys 157:060901
Van den Berg FTM, Schonenberg JHM, Beijerinck HCW (1987) Ionization of small molecules by state selected Ne*(3P0, 3P2) metastable atoms in the 0.06<E<6 eV energy range. Chem Phys 115:359–379
Weinhold F (2023) “Noncovalent interaction”: a chemical misnomer that inhibits proper understanding of hydrogen bonding, rotation barriers, and other topics. Molecules 28:3776
Willitsch S (2017) Chemistry with controlled ions. Adv Chem Phys 162:307
Zou J, Osterwalder A (2020) Investigation of the low energy stereodynamics in the Ne(3P2) + N2, CO reactions. J ChemPhys 153:104306
Zou J, Gordon SDS, Tanteri S, Osterwalder A (2018) Stereodynamics of Ne(3P2) reacting with Ar, Kr, Xe, and N2. J Chem Phys 148:164310
Acknowledgements
This paper is dedicated to the memory of Professors Gian Gualberto Volpi, Giorgio Liuti and Roberto Candori.
Funding
Open access funding provided by Università degli Studi di Perugia within the CRUI-CARE Agreement.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This peer-reviewed paper belongs to the Topical Collection originated from contributions to the conference held in Rome, March 27–28, 2023, promoted by Accademia Nazionale dei Lincei and Fondazione Guido Donegani on Chemical Kinetics at Micro-, Meso-, Bioscales, dedicated to Giangualberto Volpi (1928–2017), Linceo from1994.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pirani, F., Falcinelli, S., Vecchiocattivi, F. et al. Intermolecular interactions and the weakly bound precursor states of elementary physicochemical processes. Rend. Fis. Acc. Lincei 34, 983–995 (2023). https://doi.org/10.1007/s12210-023-01204-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12210-023-01204-x