
Abstract
In recent years, enormous efforts have been devoted to alleviating global energy demand and the climate crisis. This has instigated the search for alternative energy sources with a reduced carbon footprint. Catalytic hydrogenation of CO2 to CH4, known as the methanation reaction, is a pathway to utilise CO2 and renewable hydrogen simultaneously. However, owing to the high stability of CO2 and thermodynamic limitations at higher temperatures, the methanation process is energy intensive. Non-thermal plasma technology has recently emerged as a promising approach to lowering the activation temperature of CO2. The application of a plasma coupled with catalytic materials allows the methanation reaction to occur at or near ambient conditions, with dielectric barrier discharges providing superior performance. The review considers the various catalytic materials applied for plasma-assisted catalytic CO2 methanation and assesses CO2 conversion, CH4 yield and fuel production efficiency obtained. The importance of reactor designs and process parameters are discussed in detail. The possible reaction pathways are considered based on in-situ and other diagnostics and modelling studies. Finally, a perspective on current barriers and opportunities for advances in non-thermal plasma technology for CO2 methanation is presented.
Similar content being viewed by others

Introduction
Catalytic CO2 hydrogenation to methane, known as the Sabatier or methanation reaction (Eq. 1), is an important industrial process that is thermodynamically favourable (∆H298 K = − 164 kJ/mol) at low temperatures [1]. However, the reduction of the fully oxidised carbon to methane is an eight-electron process with significant kinetic limitations. It normally requires a catalyst to achieve acceptable rates and selectivity in the thermal process [2]. In addition, as the reverse water–gas shift (RWGS) reaction (Eq. 2) proceeds at high temperatures (above 450 °C), the production of CO increases with temperature while CH4 selectivity is reduced.
According to Gao et al. [3], CO2 methanation is the only CO2 hydrogenation reaction that can theoretically achieve 100% CO2 conversion and CH4 selectivity at temperatures < 200 °C, as shown in Fig. 1. However, converting CO2 into methane with an acceptable reaction rate and selectivity at low temperatures is inherently difficult due to kinetic limitations. Many researchers have endeavoured to find highly active catalysts to overcome the kinetic energy barrier at low temperatures [3,4,5,6,7,8]. The improvement so far is limited. Temperatures of at least 200 °C, and typically much higher, are required, necessitating elevated pressures to maintain acceptable CO2 conversion.

Effect of temperature and pressure on CO2 methanation based on thermodynamics: a CO2 conversion b CH4 selectivity. Reprinted from [3], Copyright (2021), with permission from Elsevier
Plasmas are ionized gases that contain various activated species, i.e., ions, electrons, atoms, radicals and excited molecules. Both high-energy electrons and reactive species contribute to the initiation and propagation of a variety of physical and chemical reactions [9, 10]. They are known to invoke the vibrational excitation of CO2, which facilitates its easier dissociation at a lower temperature on the catalyst surface and can activate CO2 beyond the thermodynamic limitations [11]. Plasmas of industrial interest include thermal plasmas, in which the electrons and heavy species are in thermal equilibrium, and a wide range of non-thermal plasmas (NTPs), in which the heavy species are at lower temperatures than that of the electrons. NTPs, which are also referred to as non-equilibrium plasmas, can be further divided into low-temperature or cold plasmas, in which the heavy species are close to room temperature, and warm plasmas, where the heavy species may be at 1000 K or higher [12]. NTPs may be formed at atmospheric or low pressures [13], offering an attractive alternative to the thermal catalytic route for CO2 conversion [14].
When an applied electric field is high enough to cause the electrical breakdown of a gas, the produced electrons rapidly accelerate in the electric field, typically reaching energies of 1 to 10 eV. Collisions of electrons with heavy species may result in ionization, excitation or dissociation [15]. The transfer of kinetic energy from electrons to heavy species is limited by the large mass difference. Nevertheless, at atmospheric pressure, the collision frequency is high enough to lead to thermal equilibrium within 1 μs. By interrupting the discharge [16], for example, by using a dielectric barrier interposed between electrodes, the heavy species temperature can be kept low while maintaining significant densities of excited molecules, radicals and ions [17, 18]. The excitation may help overcome the energy barriers of chemical reactions, allowing highly endothermic reactions to occur at a relatively low temperature [19, 20]. The high reaction rate and rapid attainment of steady state in a plasma system allow rapid start-up and shutdown of plasma processes compared to thermal processes. This significantly reduces the overall energy cost [14] and suits plasma devices to be coupled with intermittent energy sources such as wind or solar power.
There are many ways to produce NTPs, including DC and AC glow discharges (GDs), radio frequency (RF) discharges, microwave (MW) discharges, dielectric barrier discharges (DBDs), gliding arcs (GAs), nanosecond-pulsed discharges, and corona and spark discharges, [21, 22]. Their particular characteristics suit them for different applications [23,24,25,26,27,28].
For conversion of CO2 to other gases, the most commonly used plasmas are DBD [29], MW and GA [22], which are illustrated in Fig. 2, and their representative properties are given in Table 1.

Schematic illustration of the three different plasma reactors most often used for gas conversion applications: a DBD, b MW plasma, c GA discharge in classical configuration, and d gliding arc plasmatron. Reprinted with permission from [25]. Copyright (2018) American Chemical Society
A DBD (Fig. 2a) is created by applying an electric field between two electrodes, of which at least one is covered by a dielectric barrier, while gas is flowing through the middle. A DBD usually operates at atmospheric pressure and (near) room temperature. DBDs are used commercially for ozone production, demonstrating that scale-up is feasible. The low gas temperature and the reactor geometry allow coupling of catalyst materials in the reactor, which can be leveraged to better control conversion and the selective production of value-added compounds [32, 33].
A MW plasma is produced by applying MW power to a quartz or ceramic tube filled with gas (Fig. 2b). MW plasmas have been operated at pressures from a few mbar up to 1 bar, with better CO2 conversion and corresponding energy efficiency at reduced pressure (ca. 100–200 mbar) for CO2 splitting than at atmospheric pressure. The gas temperature can rise to above 3000 K at (sub)atmospheric pressure, which makes coupling with catalyst materials difficult. If catalyst materials are used, they are placed downstream of the plasma reactor (so called “post-plasma catalysis”).
In a GA plasma, an electric potential difference is applied between two flat diverging electrodes (see Fig. 2c); the arc propagates upward. A GA plasma typically operates at atmospheric pressure and exhibits quite good energy efficiency (Table 1). However, the conversion is limited (typically about 10%) because of the limited residence time of the gas inside the arc plasma. Similarly, the gas temperature in microwave plasmas is typically too high for catalyst materials to be placed in the plasma. To improve the interaction of the plasma and gas, three-dimensional configurations of gliding arcs, called gliding arc plasmatrons, in which the arc rotates as well as propagating upward, have been developed.
DBD plasmas have several attractive features for CO2 methanation. The suitability for scale-up has already been noted. The low gas temperature characteristic of the process offers two benefits. First, the low gas temperature thermodynamically favours a high level of CO2 conversion and CH4 selectivity (see Fig. 1). Second, the low temperature allows catalyst materials to be coupled within the plasma, facilitating synergetic interactions between the species produced by the plasma and the catalyst. Interactions of excited species with the catalyst are of critical importance for reactions with a range of potential products, including CO2 methanation, since selectivity is poor without the use of a catalyst. The short lifetimes of the relevant excited species mean that post-plasma catalysis is of limited value. Accordingly, our focus will be on DBD plasmas.
Plasma catalysis is a topical field of research that is being explored for a wide range of reactions, including ammonia production [34, 35], dry reforming of methane [36] and CO2 hydrogenation [37]. There are complex two-way interactions between the plasma and the catalyst. The presence of the catalyst in the plasma enhances the electric field intensity and affects the discharge properties. Conversely, the plasma can modify the catalyst surface. The short-lived excited species produced in the plasma, including vibrationally and electronically excited molecules, radicals and ions, allow catalyst activation at lower temperatures than ground-state molecules, making CO2 methanation possible at low temperatures, for example. The co-interactions of the plasma and catalyst create new reaction pathways. However, understanding the reaction mechanisms and their dependence on both the plasma and the catalyst is difficult. The long-term goal of developing an energy-efficient reactor system that can provide, in the case of CO2 methanation, high CO2 conversion and CH4 selectivity requires detailed planning, study and research.
The extensive studies of plasma-based CO2-conversion technologies, especially CO2 splitting and dry reforming of methane, including the influence of various parameters (plasma sources, packing materials, etc.), have been considered in reviews by Snoeckx [38] and Bogaerts et al. [22, 25, 30, 38, 39]. CO2 hydrogenation using plasma, including CO2 methanation, forms a relatively small subset of these technologies [14, 40]. With the supply of hydrogen via water electrolysis and other carbon–neutral technologies becoming a more economically viable option, plasma-catalytic CO2 methanation is attracting increasing attention because of its suitability for renewable energy storage. While an excellent review of the topic by Dębek et al. was published in 2019 [11], their focus was on the choice of catalyst. Our review updates this work and provides, in addition, detailed considerations of the diagnostic, mechanistic and modelling studies and an assessment of the importance of DBD parameters, including voltage, packing material and electrode design.
The review is divided into several sections. In the next section, we consider the characteristics of DBD reactors. The following two sections consider plasma-assisted CO2 methanation studies performed without and with catalysts, respectively; in the latter case, the individual roles of the catalytic metal, supports and promoters are reported. We devote the following section to an analysis of the performance reported in previous studies, particularly the fuel production efficiency, which we argue is the most suitable measure of energy efficiency. The subsequent section considers the role of operational parameters (voltage, frequency, packing materials, electrode material and geometry and diluent gas). We then assess the reaction mechanisms underlying plasma-assisted CO2 methanation, devoting sections to the results of experimental investigations, particularly those using in-situ diagnostics and modelling studies. Finally, the prospects for further improvements are considered in the concluding section.
Plasma DBD CO2 Methanation and Reactor Configurations
The DBD is one of the most common types of NTP for CO2 conversion. It has been known for more than a century; the first experimental investigations were reported by Siemens in 1857 for ozone generation [41].
A DBD consists of two plane-parallel or concentric metal electrodes, with at least one dielectric barrier in between the electrodes [41, 42]. The purpose of the dielectric barrier is to restrict the electric current to prevent the formation of sparks and/or arcs. The typical dielectric materials include quartz, glass, ceramics and PTFE. A gas flow is applied in the discharge gap, which has a typical width ranging from less than 0.1 mm to several centimetres, depending on the application [41]. An alternating voltage with an amplitude of 1–100 kV and a frequency of a few Hz to MHz is applied across the electrodes [13]. At atmospheric pressure, a large number of short-lived micro-discharges are usually formed as the voltage approaches its positive or negative peak values and exceeds the breakdown voltage. The micro-discharges are interrupted when the charge transferred to the dielectric reduces the electric field to below the level required to sustain the plasma.
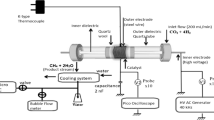
Some typical DBD plasma configurations used for CO2 hydrogenation reactions based on planar and cylindrical geometry are shown in Fig. 3 [41, 43]. For the discharge operation, one or more dielectric layers are needed between the metal electrodes. The dielectric layer can cover one or both electrodes or lie between them without contact. When both electrodes are covered with dielectric material, as shown in Fig. 3a, the discharge is called full dielectric barrier discharge. If only one electrode is covered by a dielectric layer, the discharge is called a half dielectric barrier discharge, as in Fig. 3b. The interaction of the plasma with an electrode may cause sputtering, and corrosive gases may also react with the electrode, potentially producing impurities in the plasma. Therefore, a full dielectric barrier discharge may be required for the formation of a high-purity plasma. The cylindrical DBD plasma consists of a central electrode coaxially surrounded by another electrode with at least one dielectric tube between them; Fig. 3d. This setup allows the discharge space to be coupled with catalytic materials for gaseous conversion reactions [44]. In general, DBDs operate at approximately atmospheric pressure (0.1–10 atm, but usually 1 atm), enabling gas-phase reactions at ambient conditions. We will focus on the cylindrical DBD reactor as this is the most widely studied system for CO2 methanation. A typical experimental set up for plasma CO2 methanation is shown in Fig. 4.


A typical cylindrical DBD reactor experimental set up for plasma catalysis
DBD CO2 Methanation Without a Catalyst
As mentioned previously, the non-equilibrium characteristics of a DBD plasma can overcome the kinetic barriers of chemical reactions, producing an abundance of highly active species through primary and secondary collisions, which help to create new compounds and enable highly endothermic reactions to occur at a relatively low temperature.
Research on DBDs has focused on tailoring micro-discharge characteristics by making use of special gas properties, adjusting pressure and temperature, and optimising the electrode geometry as well as the properties of the dielectric(s) [41]. The reduced electric field (the electric field normalised to the total number density) in a DBD reactor is typically above 100–200 Td, where 1 Td is 10−21 V m2, creating electrons with higher energy which mainly give rise to electronic excitation, ionization, and dissociation with electronic excitation being the dominant process (see Fig. 5). Electronic excitation to a dissociation level requires 7–10 eV, which is much more than the C=O bond dissociation energy of 5.5 eV [25]. CO2 dissociation to CO in a DBD is not regarded as energy efficient because the energy is consumed to split CO2 but not to produce CH4. The dissipated energy results in an increase in gas temperature (temperatures between 100 and 200 °C are usually reported). Bogaerts et al. [38] note that stepwise vibrational excitation, which is achievable using MW or GA discharges because of their lower reduced electric field (see Fig. 5), can be used to dissociate CO2 without expending more than 5.5 eV. Accordingly, such reactors are often used for CO2 dissociation, which does not require a catalyst. As discussed in the introduction, their application to CO2 methanation is limited.

The fraction of electron energy transferred to different modes of excitation of CO2 as a function of the reduced electric field in a CO2 plasma. Typical E/N ranges for MW, GA and DBD reactors are also shown. Reproduced from [38]. CC BY 3.0
The reported CO2 conversion to methane obtained in DBD systems without catalysts is usually below 20%, with CO being the main product (with selectivity at the level of ca. 90%) [14, 45,46,47,48,49,50]. The addition of an inert gas to make use of the Penning effect [51], increasing the CO2/H2 ratio and gas retention time, adding dielectric materials [52] and increasing input plasma power may improve the conversion and selectivity to some degree. However, CO is still the dominant product because of the electron-impact CO2 dissociation [40]. Therefore, plasma-alone processes without a catalyst show poor conversion, selectivity and low yield for CH4 production.
DBD Reactors with Catalyst Materials
Appropriate catalysts can be highly selective to CH4 in thermal reactions, but show a very low activity at low reaction temperatures (< 200 °C, or higher, depending on the catalyst). As mentioned earlier, plasmas can provide energetic species that allow reactions, such as CO2 dissociation, to proceed at low temperatures. However, selectivity to particular products, such as CH4, is usually low due to the non-selective interactions between reactive species [53]. Coupling a plasma with the catalyst can significantly enhance CO2 methanation at low temperatures while retaining high selectivity for methane because of the strong synergetic effect between plasma and catalyst [14, 54]. Many researchers have observed that materials active in thermal methanation also perform well in plasma catalysis. Among different metals, Ni, Ru and Co-based catalysts loaded on various supports showed high CO2 conversion (70–80%) and CH4 selectivity (90–100%) in DBD reactors [53, 55].
Traditional catalysis research has been dominated by structure–activity relations, implying that catalyst optimization and engineering are paramount. In contrast, plasma catalysis allows tuning of the surface chemistry beyond structure–property relations because of the complex synergetic interactions at the interface. On the one hand, all the factors that contribute to thermal catalysis are also important in plasma catalysis. These factors are the size distribution of the catalyst particles, the physical structure and chemical properties of the catalyst and the support material. These properties will affect the thermodynamic properties, such as the adsorption energy of the reactants and kinetic properties, such as the reaction barriers and rate coefficients [54]. On the other hand, the synergistic interactions between the plasma and the catalyst surface offer potential pathways to overcome the kinetic and thermodynamic barriers at low temperatures (ambient conditions), which results in enhanced conversion, selectivity, and fuel efficiency.
Plasma Pre-treatment of Catalyst Materials
The pre-treatment of catalyst materials with a plasma is an active area of research. The highly energetic species produced in a plasma may interact with catalyst materials and result in the formation of oxygen vacancies, modification of functional groups, defects, and changes on the surface of the catalyst [21, 56]. These changes can lead to different adsorption properties, improved metal dispersion, and different acidity and basicity, which affect the kinetics of the reaction.
Plasma pre-treatment has been applied to a wide range of catalysts, including those used for thermal catalysis, photocatalysis [57,58,59] and electrocatalysis [60]. Here we focus on pre-treatments of catalysts for CO2 methanation reactions.
Jantrang et al. [61] pre-treated a Ni/TiO2 catalyst for the photothermal CO2 methanation reaction in a helium plasma. They found the surface oxygen defects were stabilised on the catalyst surface if plasma treatment was employed before reduction and passivation. Otherwise, defects couldn’t be produced. The authors believed these surface oxygen defects led to enhanced CO2 adsorption and basicity, which consequently resulted in higher CO2 methanation activity.
Pastor Pérez et al. [62] treated a Pt/CeO2 catalyst with an RF Ar plasma and compared the performance for the water gas shift reaction (CO + H2O → CO2 + H2), which may occur in the CO2 methanation process. It was found plasma pre-treatment increased the electronic density of Pt, which improved the metal support interaction, resulting in an improved reaction.
Basicity improves the adsorption and activation of CO2. The presence of medium-strength basic sites exhibited a vital role in the catalytic performance of the CO2 methanation reaction [63]. Ge et al. [64] investigated the influence of H2-plasma reduction of a Ni-Ce nanoporous catalyst. They found the catalyst reduced under plasma had almost 1.5 times more medium-strength basic sites than the catalyst reduced by the thermal method. Similar observations and conclusions about the importance of medium-strength basic sites were also reported by Pan et al. [65]
Recently, Benrabbah et al. [66] compared the effect of thermal and plasma H2 reduction of a Ni/CeZrO2 catalyst on its performance in the plasma catalytic methanation reaction. Their results demonstrated catalyst reduced under plasma under ambient conditions at an input power of 4 W gave higher CO2 conversion (73%) than the catalyst reduced under thermal (470 °C) conditions (63%); the conversion was similar for both catalysts at higher powers. Characterisation revealed that plasma treatment reduced crystallite size and enhanced the basicity of the surface. Perhaps the most striking result presented by Benrabbah et al. is shown in Fig. 6. A calcined catalyst that was not pre-treated thermally or in a plasma was found to become highly active for methanation performed at plasma powers of 12 W or more, indicating that in-situ activation occurs in plasma catalytic CO2 methanation when the power is sufficiently high. Thus, hydrogen plasma, either as a pre-treatment or applied in in-situ, can replace thermal pre-treatment.

CO2 conversion and CH4 yield against input power for plasma CO2 methanation over calcined Ni/CrZrO2 not exposed to any pre-treatment. Reprinted from [66], Copyright (2017), with permission from Elsevier
In summary, the studies so far indicate that plasma pre-treatment of catalyst material before CO2 methanation and other reactions can significantly alter the surface properties of the catalyst, such as metal support interaction, particle size, surface basicity, etc. These improved surface properties enhanced the catalytic activities of the materials.
Catalyst Materials for Plasma CO2 Methanation
As discussed previously, the plasma process is active towards CO2 hydrogenation but shows poor selectivity for CH4 when no catalyst is used. Thermal catalysis employs catalysts that are highly selective for CH4 but show very low activity at low reaction temperatures (e.g., < 150 °C). Researchers have been attempting to combine the two processes since 2011 [67]. However, the complexity of plasmas and their interactions with catalysts means that significant effort is still required to understand the fundamental chemistry at the interface and to tailor the catalyst and process conditions to optimize process efficiency [68, 69].
Catalysts usually employ a catalytic metal finally dispersed on a metal oxide support material, often with an alkali or rare-earth metal promoter incorporated in the support. While the effectiveness of the catalyst and its interactions with the plasma depend on all three components, studies usually focus on one component. Detailed insight is required into each component to understand the complex plasma chemistry; therefore, we consider the metal, support and promoter separately in the next three sub-sections.
Metals
Many researchers have observed materials active in thermal methanation also performed well in the plasma catalytic process. Ni-based catalysts are the most widely used materials for thermal CO2 methanation because of their low cost and high activity and selectivity towards CH4. However, they often experience sintering and coking at higher temperatures. The plasma process typically uses mild or even ambient temperatures. Therefore, Ni-based catalysts have been widely studied for the plasma CO2 methanation process.
Jwa et al. [70] explored the role of Ni-zeolite in plasma CO2 methanation. They confirmed that Ni dispersion was improved, and no sintering was observed during the plasma reaction. Nizio et al. explored CO2 methanation in a DBD reactor over Ni-containing hydrotalcite-derived catalysts with Ce and Zr promoters. Around 80% CO2 conversion and 9% CH4 selectivity were obtained at 90 °C. The same conversion and selectivity obtained under plasma conditions were achieved at 300 °C in the thermal-only reaction. For comparison, the plasma-only test was performed without a catalyst at 90 °C; CO2 conversion was reduced to around 5%, with CH4 and CO yields of 0% and 5%, respectively [49]. No evidence was found of any morphological modification of the catalysts or their basicity after 100 h exposure to the plasma.
The selective role of Ni, Mn, Fe and Co over a ZSM-5 support was investigated by Lan et al. [69] in CO2 hydrogenation in a DBD plasma reactor. The findings revealed that product selectivity was highly reliant on the active metal involved. Ni, Mn and Fe on ZSM produced methane. The maximum CO2 conversion of 46.3% was obtained for the Ni/ZSM-5 catalyst, while Co/ZSM-5 produced the most C2-C4 hydrocarbons.
Zeng et al. [14] compared Cu/Al2O3, Mn/Al2O3 and Cu-Mn/Al2O3 for CO2 hydrogenation and found CO2 conversion was enhanced by 6.7–36% when plasma was coupled with these materials compared to the plasma-only process. Mn was found to be the most active metal for the CO2 hydrogenation process, and Cu was the least active metal. Cu-based materials are active for the water–gas shift (WGS) reaction that converts CO and H2O to CO and H2, so the presence of Cu might promote this back reaction, leading to reduced CO2 conversion. The CH4 selectivity was less than 10% for all the catalysts, indicating that the metals are not selective for methanation.
In a detailed study of Co, Wang et al. [71] examined the synergistic effects between the DBD plasma and a Co/Al2O3 catalyst. Their experimental results revealed the plasma lowered the activation energy for CO2 hydrogenation by 50% compared to the thermal catalytic reaction. A strong synergistic effect of the plasma and catalyst was demonstrated when comparing CH4 production with and without the Co catalyst and the plasma. Kinetic studies combined with DRIFTS (diffuse reflectance infrared Fourier-transform spectroscopy) measurements confirmed the plasma promoted CO2 activation and reduced hydrogen adsorption on the catalyst surface. Moreover, the surface-adsorbed hydrogen species were much more reactive. The CO produced in the plasma phase reacted with surface-adsorbed H, forming methane. Without the Co catalyst, the support material was not able to facilitate CH4 formation from gas-phase CO and H species. It was also found that pre-adsorbing CO2 on the catalyst surface deactivated the catalyst, with no methane being produced by reactions between gas-phase H and surface-adsorbed carbon-containing species.
Evidence of a synergistic effect between plasma and Ru/Zr-MOF (metal–organic framework) was found by Xu et al. [68]. The catalyst displayed enhanced CO2 conversion of 41.3% under plasma, almost 1.9 times greater than that found with Zr-MOF. The CH4 selectivity and yield were 94.5% and 39.1%, respectively. The improved methanation performance was ascribed to the increased reducibility of Ru3+ ions to Ru0 in the Zr-MOF pores and the stable structure of the catalyst. Xu et al. demonstrated in a recent study that the plasma could alleviate the CO poisoning effect by removing the strongly adsorbed carbon species from the catalyst surface [72].
Of the different metal catalysts explored so far for plasma-catalytic CO2 hydrogenation, it is clear the metals active for thermal methanation, such as Ni, Co and Ru, have also shown high performance for plasma methanation. On the other hand, metals that are not typical active methanation catalysts, such as Cu and Mn, also showed limited performance for plasma catalytic CO2 hydrogenation.
Support Materials
The properties of catalyst supports, such as the dielectric properties, morphology and pore structure, play a decisive role in NTP catalytic reactions. The properties can affect the interactions with the active metal, the stability of the catalyst, the dispersion of the active metal, the adsorption of reactants and the plasma discharge characteristics.
Chen et al. [73] investigated hierarchical meso-micro-porous structures with different accessibility and dispersion of active Ni sites. Their findings demonstrated the location of active sites in the porous support structure affects the catalytic activity. Specifically, at low input energy, the availability of active plasma species is limited, and they interact only with exposed Ni active sites with less diffusion resistance. At high energy input, in contrast, the abundance of reactive plasma species overcomes the diffusion resistance and enables interaction with highly dispersed active metallic sites. The increased interaction between short-lived plasma species and highly dispersed Ni sites results in an increased CO2 conversion rate of 75% with high CH4 selectivity of 95%.
The nature of the support also plays a vital role in plasma-assisted catalysis. Dębek et al. [74] explored the performance of Ni-based catalysts for CO2 methanation reaction in glow discharge plasma using Al2O3, SiO2 and CeO2-ZrO2 supports. The results showed the physicochemical properties of the support have a strong influence on the plasma properties. Ni/Al2O3 catalyst exhibited the best performance towards CH4 formation, which was ascribed to increased Ni dispersion and enhanced adsorption for CO2 under plasma. In contrast, Ni/SiO2 was found to be inactive under a glow discharge plasma, with no CH4 produced. An analysis of the mechanism revealed that CO adsorption was the vital step for CH4 formation, and since no sites were available for CO adsorption, CH4 was not produced. In the case of the Ni/Ce-Zr catalyst, CO adsorption capacity was increased, but the Ni sites were oxidised, reducing the performance.
MOFs as support materials have exceptionally high surface areas and abundant surface hydroxyl groups, which help the dispersion of active metals. Additionally, MOFs have stronger CO2 adsorption capability than zeolite materials, which can be useful for CO2 hydrogenation reactions. To test the stability of MOFs, Chen et al. [75] investigated the Ni/UiO-66 catalytic material under NTP. They found after 20 h testing, the turnover frequency (TOF) of the plasma-assisted system was three times that of the thermal system (1.8 s−1 vs. 0.06 s−1). The structure of the catalyst material was also found to be stable in the NTP. Chen et al. also found that the performance of the Ni/UiO-66 catalyst exceeded that of other supported catalysts (Ni/ZrO2 and Ni/α-Al2O3). The superior performance of the MOF-based catalyst was attributed to the formation of new species under the influence of the NTP that were absent for the ZrO2 and Al2O3 supports.
Bacariza et al. [45] investigated the effect of the Si/Al ratio and Ce addition in Ni/USY zeolite catalysts. They found the performance of the catalyst was linked to the dielectric properties of the support and its affinity to water. A lower Si/Al ratio increased the dielectric constant of the support material. While this was expected to enhance the electric field and charge accumulation on the catalyst surface, affecting the plasma composition, it was concluded that this was not the most important factor in determining catalytic performance. A more significant effect was the reduction in the catalyst’s affinity to the water produced by the methanation reaction with increasing Si/Al ratio, providing more active sites for the methanation reaction. Further, the addition of CeO2 enhanced the catalytic performance towards methanation reaction under plasma conditions, which was attributed to increased dielectric constant of CeO2 (24) compared to zeolite materials (1.5–5). The influence of the Cs promoter was also found to be significant; this is considered in the next sub-section.
Promoters
The addition of promoters such as alkali and rare-earth metal oxides could improve the reducibility and dispersion of the active metal and its interactions with the support material in various catalytic systems. In their study discussed in the previous section, Bacariza et al. [45] investigated the influence of Ce as a promoter in Ni/USY zeolite catalysts. They found that Ce, on the one hand, improved the active Ni metal dispersion on the support, providing additional sites for CO2 activation. On the other hand, it increased the dielectric constant of the catalyst, which was hypothesised to promote CO2 activation. An overall best result of 66% CO2 conversion and 97% CH4 selectivity was achieved at 20 W. Similar results and conclusions were obtained by Amouroux and Cavadias [76] using an Ni/SBA-15/CeZrO2 catalyst, achieving up to 80% CO2 conversion and 100% CH4 selectivity.
The effect of CeO2 content in the range of 0–50% on a Ni/Al2O3 catalyst was explored by Andreu et al. [77]. They found CeO2 addition was beneficial for both thermal and plasma catalytic processes. However, lower CeO2 content (10 wt%) was preferred for plasma conditions, while higher CeO2 content (40 wt%) was found to be optimal for the thermal reaction. The improved performance under plasma with lower CeO2 content was thought to originate from CO formation in the gas phase, but details of the underlying mechanism were not explored.
The use of promoters such as Co, Cu, Mn, La, Y, Gd and Sr was also explored for Ni catalysts [63, 78]. These promoters encompass alkaline earth metals, transition metals, lanthanide metals and rare-earth metals, spanning a wide range of dielectric constants and bandgaps. The presence of these elements was found to considerably alter the physical, chemical and electrical features of the catalysts, including the distribution of the basic sites, the dispersion of the active metal and the support crystallite sizes. The best catalysts often had a high percentage of medium-strength basic sites and high catalyst dispersion, which is in accordance with thermal methanation results.
The individual effects of Ce and Zr as promoters were explored by Nizio et al. [50] for Ni-containing hydrotalcite-derived catalysts. They found the addition of Ce or Zr to the support produced Lewis metal oxygen pairs and strong Lewis-base oxygen anions, which increased the total basicity of the catalyst. 80% CO2 conversion and 100% CH4 selectivity were observed when the catalyst was coupled with a plasma. The authors hypothesised that low or medium-strength basic sites helped to boost the methanation process while strong sites were not beneficial, in agreement with the understanding of the thermal CO2 methanation process.
Hasrack et al. [78] examined the promotional role of 1% and 5% Co on Ni/CeO2 catalysts. They found Co addition increased the number of basic sites, especially the medium-strength basic sites. The catalyst with 1% Co displayed the highest number of medium-strength basic sites and exhibited the best performance under plasma and thermal conditions. Similar observations were presented by Ge et al. [64] and Wierzbicki et al. [79]. Pan et al. [65] provided further evidence for the promotional effect of medium-strength basic sites. They found that monodentate formate derived from monodentate carbonate on medium-strength basic sites was hydrogenated quickly to methane. In contrast, the strong basic sites didn’t participate in the methanation reaction.
The research conducted so far on different catalytic materials has shown the catalysts active for thermal methanation also showed enhanced performance under NTP. The physical interactions between plasma and catalysts vary for different metals. Among the active metals, Ni-based catalytic materials displayed enhanced CO2 conversion and CH4 selectivity for plasma-assisted CO2 methanation. The plasma properties are strongly dependent on the nature of the support, and, as discussed in the “Dielectric Packing Materials” sub-section, the dielectric constant of the support affects the discharge characteristics. The addition of promoters also plays a key role in controlling the properties of catalytic materials under plasma conditions. For instance, promoters can enhance the percentage of medium basic sites on the catalyst surface, which is one of the determining factors in CO2 methanation.
Energy Efficiency of DBD Plasma CO2 Methanation for Different Catalyst Systems
As discussed previously, the energy efficiency of the plasma CO2 methanation process is a critical parameter in determining its industrial applicability. While features of the plasma process, such as the ability to operate at lower temperatures, provide advantages over thermal CO2 methanation, the energy efficiency has to be competitive. In this section, we use literature data obtained on different catalyst systems to examine the energy efficiency of the plasma catalytic process.
We first present a summary of the main performance indicators used for plasma CO2 methanation is presented.
Specific Energy Input and Energy Cost
Specific energy input, abbreviated as SEI, is defined as the ratio of power supplied to the gas for plasma generation and the input flow rate of the gas. This factor is critical in determining the energy efficiency of the plasma system. Typical units are kJ/L or eV/molecule.
where Pd is the discharge power and rin is the input gas flow rate.
The conversion to eV/molecule is given by
where the far-right-hand equality is calculated using the molar volume Vmol = 24.5 L/mol that applies at normal temperature and pressure.
The energy cost (EC) is the amount of power consumed per mole of CH4 formed:
where \(r_{{\text{CH}}_4 ,{\text{out}}}\) is the molar of production of CH4. The CO2 conversion in the gas mixture is defined as
where \(n_{{\text{CO}}_2 ,{\text{in}}}\) and \(n_{{\text{CO}}_2 ,{\text{out}}}\) are the number of moles of CO2 in the inlet and outlet of the plasma reactor, respectively.
The selectivity for CH4 is given by:
where \(n_{{\text{CH}}_4 ,{\text{out}}}\) is the number of moles of CH4 in the outlet of the plasma reactor.
The yield of CH4 is obtained by multiplying its selectivity by the conversion of the CO2
The conversion, selectivity and yield can also be calculated from of flow rates, since, for example, \(r_{{\text{CH}}_4 ,{\text{out}}} = n_{{\text{CH}}_4 ,{\text{out}}} /\Delta t\), where Δt is the time over which the process is run.
Energy Efficiency Calculation
The energy efficiency of plasma process is an important parameter. Various methods have been used by different researchers to calculate the energy efficiency for CO2 hydrogenation processes.
-
(1)
Equation adopted from CO2 splitting process
The energy efficiency for CO2 splitting is calculated as
where \(r_{{\text{CO}}_2 ,{\text{in}} }\) is the molar flow rate of CO2 at the inlet of reactor and \(\Delta\)HR is the reaction enthalpy, 279.8 kJ/mol for the CO2 splitting process at 298 K. Some researchers (e.g., Dębek et al. [11]) adopted an analogous formula for CO2 hydrogenation
where \(X_{{\text{total}}}\) is the total conversion, obtained by summing the products of the conversion of reactant gases with their inlet mole fraction:
Equation (9) is an appropriate expression for energy efficiency since the plasma is the only energy source. CO2 splitting is an endothermic process, so the reaction enthalpy reflects the difference between energy output and input into the process. However, CO2 hydrogenation is very different from CO2 splitting, and Eq. (10) is not a suitable measure of energy efficiency. For example, CO2 methanation is exothermic, so the reaction enthalpy is negative (–165 kJ/mol), which indicates that process heat is generated during the reaction and may be wasted if not recycled. Hence, the reaction enthalpy does not represent the effective output energy of the process. Inserting this value in Eq. (10) gives a negative energy efficiency that represents lost energy (if there is no recycling) and not the output energy of the process. Furthermore, for CO2 hydrogenation, there is another feed gas, H2, that has heating value and provides energy to the process that is not reflected in Eq. (10). Products such as CH4 and CH3OH also have heating value, but the corresponding output energy is again not considered in Eq. (10). Moreover, there is no reason that |η| in Eq. (10) is restricted to values below 100%. For these reasons, we recommend against using Eq. (10) to report the energy efficiency for CO2 hydrogenation.
-
(2)
Equation based on fuel production efficiency (FPE)
Mikhail et al. [63] and Biset-Peiró et al. [80] reported the energy efficiency of hydrogenation processes based on the overall energy input and output, considering not only the power input but also the net energy flows associated with the reactants and products. For example, the fuel production efficiency (FPE) of the plasma CO2 methanation reaction was defined as the ratio between output energy in the form of CH4 (without considering unreacted H2 in the product) and the overall energy input, as shown in Eq. (12). The outlet energy was calculated based on the lower heating value (LHV) of the CH4 product, which is 801 kJ/mol. The total input energy was the power input from the plasma and the LHV of converted H2, which is 242 kJ/mol. The FPE of the plasma CO2 methanation reaction is therefore defined as
where \(r_{{\text{H}}_2 ,{\text{in}}} - r_{{\text{H}}_2 ,{\text{out}}}\) = \(\left( {n_{{\text{H}}_2 ,{\text{in}}} - n_{{\text{H}}_2 ,{\text{out}}} } \right)/\Delta t\) is the rate of consumption of H2.
The CO2 conversion, SEI and FPE for literature studies of plasma CO2 methanation using different catalysts, calculated by the above methods, are presented in Table 2 and plotted in Fig. 7. We have also included the energy efficiency calculated using Eq. (9) for completeness, even though we deprecate its use.

Performance measures calculated for plasma CO2 methanation processes using different catalysts (squares with different colours), a representative thermal CO2 methanation process (red square with dashed circle) and the plasma process without catalyst (empty squares): a CO2 conversion as a function of SEI; b fuel production efficiency as a function of SEI; c fuel production efficiency as a function of CO2 conversion; d energy efficiency based on CO2 conversion using a reaction enthalpy of 165 kJ/mol. The data and references are given in Table 2 (Color figure online)
Figure 7a shows the relation between CO2 conversion and SEI. The collected results show the Ni-based catalyst materials outperformed the catalyst materials containing other active metals such as Co, Fe, Pd and Pt; the two exceptions are Ru/MgAl and Co/Zr-MOF. The Ni catalyst that achieved the highest CO2 conversion (86%) was 15%Ni on CeO2. Figure 7b shows the dependence on SEI of the FPE calculated based on methane production. Again, the Ni-based catalysts showed better FPE than the catalysts containing other metals in almost all cases. The Ni catalyst with the highest FPE (74.1%) consisted of 15%Ni and 30% CeO2 loaded on Al2O3. It is also worth mentioning that most of the highly efficient Ni catalysts contained CeO2 in the catalyst structure to some degree. Wang et al. [95] hypothesised that the effectiveness of CeO2 is associated with its high dielectric constant and unique oxygen storage capacity. Figure 7a and b also indicate that increasing the SEI tends to decrease the CO2 conversion and methane production process efficiency. The best FPEs were all achieved at SEIs less than 5 kJ/L; Ni catalysts were used in all these cases except for one study that used Ru supported on MgAl layered double hydroxide.
Figure 7c shows the relationship between FPE and CO2 conversion. It is clear that processes that achieved high CO2 conversions also showed high process energy efficiencies. To help understand the difference between plasma and thermal CO2 methanation in terms of fuel production efficiency, we show data for a representative thermal process [80] in Fig. 7c. The thermal process was chosen because it represents standard thermal CO2 methanation performance with a commercially available catalyst and the reference provided sufficient data for comparison purposes; it is not necessary the best published performance. Nonetheless, Fig. 7c shows that plasma CO2 methanation can provide comparable energy efficiencies to the thermal process when an appropriate catalyst is selected, and (as shown in Fig. 7b) providing the SEI is relatively low. The energy efficiency was also calculated based on Eq. (9) with \(\left| {\Delta H_R } \right| = { }\)–165 kJ/mol; the results are summarized in Fig. 7d. It is noted that some energy efficiencies values are higher than 100%. As discussed above, the fuel production efficiency (FPE) based on Eq. (12) is recommended as an appropriate measure of energy efficiency.
Plasma Process Operational Parameters
The catalytic performance in the plasma-assisted CO2 methanation strongly depends on the choice of operational parameters. Parameters such as voltage, frequency, diluent gas and reactor packing material affect the properties of plasma discharge and, so, the performance of the reactor. In this section, we will briefly explain the potential impact of these operational parameters on performance.
Voltage
The applied voltage determines the power supplied to the plasma reactor for a given arrangement and is a critical operational parameter. Increasing the voltage increases the E/N and, therefore, electron energy, promoting dissociation and ionization, as shown in Fig. 5. The discharge current also increases, with more microdischarges of higher current density formed. Hence, the discharge power increases more rapidly than the voltage.
Mikhail et al. [81] applied a range of voltages in their study of plasma-assisted CO2 methanation using Ni/CeZrO2 catalysts. The observed optimum voltage for their process was 15 to 16 kV, with 71% CO2 conversion and 95% CH4 selectivity achieved at 16 kV. In this condition, the temperature and power were found to be in the range of 230 to 270 °C and 8 to 12 W, respectively. A small decrease in voltage from 15 to 14.5 kV reduced the conversion and selectivity significantly, with CH4 selectivity dropping to 20%. The authors suggested that the lower voltage promoted the direct splitting of CO2 to CO, which became adsorbed on the catalyst surface at temperatures inadequate for its desorption and subsequent conversion to CH4. This explanation seems unlikely since the dissociation of CO2 is promoted by higher E/N. For voltages above 16 kV, the reactor temperature increased to 337 °C, indicating more energy was lost as heat, and while the conversion remained approximately constant, the product selectivity shifted towards CO via the RWGS reaction [96].
The same researchers [83] studied plasma CO2 methanation reaction as a function of applied voltage in the range of 13 to 14.5 kV over three Ni/Zr catalysts with three different promoters (lanthanum, yttrium and tungsten). Their results demonstrated that increasing the voltage increased CO2 conversion but with decreased CH4 selectivity because CO was produced at higher voltages. The best CO2 conversion was achieved at 14.5 kV and CH4 selectivity at 13 kV.
Xu et al. [90] studied the effect of applied voltage in the range of 5.5 to 7.5 kV using an Ru/MgAl LDH catalyst. The CO2 conversion and CH4 selectivity initially increased with an increase of voltage from 5.5 to 6.5 kV, reaching a maximum conversion of 85% and CH4 selectivity of 98.4% at 6.5 kV, as shown in Fig. 8. With a further increase of voltage from 6.5 kV, both CO2 conversion and selectivity started deteriorating. This slight decrease in CO2 conversion and CH4 selectivity at higher input powers could be a consequence of the occurrence of various side reactions, including direct CO2 splitting, the RWGS reaction, and reverse CO2 dissociation, i.e., recombination of CO and O in a strong ionized gas [97, 98].

Effect of input voltage/power on a CO2 conversion b CH4 selectivity for Ru/Mg–Al catalysts reduced at different temperatures; for example, R160 denotes 160 °C. Reproduced from [90]. CC BY 4.0
Bacariza et al. [45] obtained somewhat different results using zeolite-based catalysts, with CO2 conversion and CH4 selectivity found to increase with input voltage from 6 to 9 kV. They hypothesised that the lower selectivities at low input voltage occurred because there was insufficient energy to drive the hydrogenation reactions required to transform the CO formed by CO2 dissociation to CH4. This explanation doesn’t consider the role of reactions on the catalyst surface. It should also be noted that the CO2 conversion is low (< 20%) for selectivities below 90%.
Mok et al. [99] studied the conversion of CO to CH4 and investigated the effect of different applied voltages (7.7, 9.0, 10.1 kV), keeping all other parameters constant. They found the voltage of 7.7 kV promoted the CO conversion and CH4 selectivity. However, when the voltage was increased to 9 kV, the conversion and selectivity did not increase proportionally as expected but only slightly, which was ascribed to the side reactions discussed above.
The results indicate that the optimum voltage depends on the reactor configuration, including the catalyst materials. In all cases, there is a threshold voltage required to obtain high conversion and selectivity, as per the example shown in Fig. 8. In some cases, the conversion and selectivity begin to decrease as the voltage is increased further, while in others, they continue to increase. It is important to note that the discharge power increases more rapidly than the CH4 production rate above the threshold voltage, so the energy cost increases with voltage. The choice of voltage for a particular system will be based on a balance between conversion, selectivity, and energy cost considerations.
Discharge Frequency
The discharge frequency is another important parameter that affects ionisation, radical formation and excitation of molecules in plasma CO2 hydrogenation reactions [46, 100]. With increasing frequency, the density of electrons and ions in the plasma increases; this is a consequence of a decreasing breakdown voltage associated with charging of the dielectric during the previous half-cycle. [101]. In addition to affecting the plasma composition, the plasma species interact with molecules on the catalyst surface.
Some studies were performed in DBDs without catalysts. Kano et al. [102] investigated the influence of a radiofrequency (RF) impulse discharge on the CO2 hydrogenation reaction in the range of 10–60 kHz in a low-pressure (1–10 torr) DBD, detecting CO, H2O, CH4 and CH3OH as reaction products. They found CH4 production increased with increasing frequency from close to zero at 10 kHz, while CO production decreased with increasing frequency. The authors explained the trend based on the hypothesis that the electron density ne increased proportionally with frequency. As CO2 dissociation to CO proceeds through electron impact, CO production will increase with ne. On the other hand, CH4 production proceeds via multiple collisions between the CO and H reactions, so its concentration will depend on nCH4 ∝ neN where N \(\ge 2\), so the CH4 will replace CO as frequency increases. A similar effect of pulsed frequency was observed by Song et al. [103, 104] for CO2 reforming of methane and Jahanmiri et al. [103, 104] for naphtha cracking in atmospheric-pressure DBDs without catalysts, where the increase in frequency led to a proportional increase in discharge power the overall conversion of the process.
When a catalyst is used, the effect of frequency becomes more complicated. Men et al. [105] investigated CO2 hydrogenation to produce methanol in a DBD with Pt catalysts for frequencies from 10 to 30 kHz. Conversion and selectivity reached a maximum at 20 kHz, as shown in Fig. 9a. The discharge power increased with frequency, promoting the CO2 hydrogenation reactions. The authors attributed this to increased electron energy, but increased electron density is a more reasonable explanation. The decreased conversion and selectivity at high frequencies are likely due to the dissociation of the product species through reactions with electrons. This is consistent with the modelling results of van‘t Veer et al., who showed that ammonia was produced by surface reactions between microdischarges, and dissociated by electron-impact reactions during microdischarges, in plasma catalytic ammonia production [106].

a Effect of discharge frequency on a CO2 conversion XCO2, methanol selectivity \({\text{S}}_{{\text{CH}}_3{\text{OH}}}\) and methanol yield \({\text{STY}}_{{\text{CH}}_3{\text{OH}}}\) for a DBD with a Pt/film/In2O3 catalyst. Reprinted from [105], Copyright (2019), with permission from Elsevier. b CO2 conversion and CH4 selectivity for a DBD with a Ru/Al2O3 catalyst. Reprinted from [46], Copyright (2017), with permission from Elsevier
Lee et al. [46] investigated the effect of frequency on CO2 methanation over Ru/γ-Al2O3 in the DBD reactor in the lower frequency range of 1–3 kHz at a fixed applied voltage of 9 kV. Their work demonstrated the conversion of CO2 and CH4 selectivity gradually increased to 10% and 85%, respectively, as the frequency increased from 1 to 3 kHz, as shown in Fig. 9b, with a sharp increase in the CH4 selectivity when the frequency increased from 2.5 to 3 kHz, which was suggested to be associated with the interactions of plasma species with the of the catalyst.
While results indicated the influence of frequency in DBDs without catalysts is dominated by the role of the increase in electron density with frequency, the presence of a catalyst complicates the interactions since surface reactions are not directly dependent on the electron density. This leads to a less direct dependence of the product concentrations on frequency.
Dielectric Packing Materials
We have already highlighted that plasma-assisted CO2 methanation has very low energy efficiency, CO2 conversion and CH4 selectivity when a catalyst is not used; see, for example, Fig. 7. Including a catalyst necessitates the use of dielectric packing materials in the reactor as catalyst supports. Dielectric materials such as quartz, glass beads, BaTiO3, TiO2, Al2O3 and ZrO2 are also often packed together with the catalysts in the discharge zone of the DBD plasma reactors [107, 108]. The presence of these packing materials enhances the electric field in the gaps between the packing material since polarisation reduces the electric field in the packing material, requiring an increased field in the gaps to maintain the applied voltage. The increased electric field results in higher mean electron energies. The packing materials also affect the physical characteristics of the discharges, for example, promoting the formation of surface discharges in addition to the filamentary discharges present in empty DBDs [109]. For example, Mei et al. [110] showed that packing glass and BaTiO3 beads (with respective dielectric constants of 3.9 and 10,000) in a cylindrical DBD enhanced the electric field by factors of 1.5 and 2, respectively, as shown in Fig. 10a, and the mean electron energy by similar factors for the same applied power.

a Effect of packing materials on electric field strength as a function of different discharge powers. Reproduced from [110], © IOP Publishing Ltd. All rights reserved. b Energy efficiency for CO2 conversion of 63% for the catalysts listed in Table 3. Reprinted from [63], Copyright (2021), with permission from Elsevier
Mikhail et al. [63] investigated a DBD packed with NiCZ-M (nickel-ceria-zirconia-metal) catalysts for CO2 methanation. The catalyst particles were spherical with 30 μm grain size; the metal M acts as a promoter. Table 3 shows that the breakdown voltage decreases with increasing dielectric constant of the catalyst; this is a consequence of the increased electric field in the gas region between grains, as is the increase in the total charge. Further, the magnitude of microdischarges increased, resulting in an increased leakage current density and power consumption. There is no clear trend in CO2 conversion or CH4 selectivity; this is more strongly affected by other properties of the catalyst. As a consequence, the energy cost of CH4 production tends to increase with the dielectric constant, as shown in Fig. 10b.
The size of the dielectric packing materials in a DBD reactor can also influence the plasma properties. Michielsen et al. [108] compared the effect of glass wool and spherical beads of SiO2, ZrO2, Al2O3 and BaTiO3 with diameters ranging from 1.25 to 2.24 mm for CO2 splitting to form CO and O2. Of the different packing materials, BaTiO3 beads had the highest dielectric constant and showed the highest conversion (25%) and energy efficiency (4.5%). The larger BaTiO3 beads, of size 2.0–2.4 nm, produced higher CO2 conversion and energy efficiency because they generated a stronger electric field and higher electron density. Zhang et al. [111] obtained similar results. It is important to note that the factors determining energy efficiency for CO2 splitting differ from those for CO2 methanation. For CO2 methanation, the increased electron energies associated with higher dielectric constant packing materials lead to increased energy cost since energy is coupled to dissociation and ionization rather than excitation of molecules.
Electrode Material and Geometry
The materials used for DBD electrodes can affect the discharge through their electrical and thermal properties and surface morphology; if the electrode contacts the plasma, it can also affect the discharge chemistry.
For example, Scapinello et al. [112] investigated the catalytic effect of different metallic electrodes, including copper, nickel and stainless steel, in a DBD reactor for CO2 hydrogenation. The authors reported the selectivity towards end products was affected when the electrode material was changed to Cu or Ni from stainless steel, indicating the importance of the electrode material.
The electrode configuration can also improve the selectivity of a targeted product. Wang et al. [113] explored three plasma reactors with different ground electrodes for CO2 hydrogenation to methanol: a cylindrical reactor (Al foil as a ground electrode, reactor 1), double DBD reactor (water as a ground electrode, high-voltage electrode covered with quartz, reactor 2) and single DBD reactor (water as a ground electrode, reactor 3). When operated without a catalyst, the three reactors gave similar CO2 conversion, but reactor 1 produced mainly CO and almost no methanol, while reactors 2 and 3 had approximately 30% and 54% selectivity for methanol, respectively. The temperature in reactor 1 increased to 350 °C because of the exothermic nature of the reaction, while the use of water as a ground electrode allowed the temperature to be maintained at 30 °C, which favoured methanol production. The quartz dielectric surrounding the high-voltage electrode in reactor 2 gave a more uniform discharge; the more filamentary discharge in reactor 3 also appeared to favour methanol production.
Studies of CO2 splitting have also demonstrated the influence of electrode properties. While the requirements for CO2 splitting are different from those for CO2 methanation, the results illustrate further ways in which electrode properties can affect DBDs. Mei et al. [114] compared CO2 conversion and energy efficiency for stainless-steel rod and screw-type high-voltage electrodes, and aluminium foil and stainless steel mesh ground electrodes. The best results were obtained for the screw-type stainless steel and aluminium foil electrodes. The superior performance was attributed to the increased electric fields near the sharp edges, the screw-type electrode and the improved effective discharge area produced by a foil compared to a mesh electrode. Both these enhancements increased the electric charge amplitude.
Lu et al. [115] investigated the impact of the shape of the inner high-voltage electrode in CO2 splitting. They compared three different configurations: a stainless steel rod, stainless steel coil and three different conductive powders, iron, copper and carbon. The copper powder electrode gave the highest CO2 conversion and energy efficiency value, which was attributed to its high electrical conductivity, resulting in a stronger electrical field in the discharge zone. Wu et al. [116] compared copper, aluminium, and stainless steel high-voltage electrodes. For the endothermic CO2 splitting reaction, high temperatures are beneficial, so low thermal conductivity is favoured. It was found that the aluminium electrode performed best because it provided the best balance between low thermal conductivity and high electrical conductivity. It should be noted that high thermal conductivity is expected to be beneficial for CO2 hydrogenation since the reactions are exothermic.
Diluent Gas
Diluent gases are often added to DBDs to alter the reaction kinetics and the discharge characteristics. Argon, which has been used in CO2 methanation, has a lower breakdown voltage than CO2 due to its higher Townsend ionisation coefficient. Therefore, Ar ionises earlier and provides more electrons to dissociate CO2, improving the conversion of the plasma processes [51, 117].
Zeng et al. [51] investigated the effect of argon as a diluent in the CO2/H2 feed gas for CO2 methanation with an Ni/Al2O3 catalyst. The CO2 conversion and CH4 selectivity increased as the Ar concentration increased from 0 to 60%. The presence of Ar decreased the breakdown voltage of feed gas and improved the charge transfer from the dielectric layer. Interestingly, the CH4 selectivity increased with Ar concentration, while the CO selectivity remained approximately constant. It was postulated that the presence of metastable excited Ar atoms, which provide new reaction pathways for the dissociation of CO2 and H2, promoted CH4 formation.
Lee et al. [46] compared the effects of the addition of N2 and equal quantities of N2 and Ar to the CO2/H2 feed gas in a DBD with a Ru/γ-Al2O3 catalyst. The presence of Ar in the diluent gas increased CO2 conversion despite the 60% higher total flow rate. The selectivity for both CH4 and CO was increased, indicating simultaneous enhancement of both methanation and deoxygenation processes. This was postulated to be a result of increased concentrations of active species.
Experimental Investigations of Reaction Mechanisms
In thermal catalytic CO2 methanation, the reactant adsorption and reaction processes follow the typical L–H (Langmuir–Hinshelwood) mechanism. The reactions are usually classified into three routes [118]: (1) conversion of CO2 to carbonates prior to methanation, CO2 → CO → OCH2 → OCH3 → CH4 (mechanism with CO as intermediate); (2) conversion of CO2 to adsorbed CO2* to formate prior to methanation, CO2* → HCOO* → CHx* → CH4 (mechanism with CO2* as intermediate); (3) direct hydrogenation of CO2 to CH4, CO2 → C + 2O + 4H → CH2 + 2H → CH4; see Fig. 11a. Thermal catalytic CO2 methanation is constrained by thermodynamic and kinetic limitations.

a Proposed mechanisms for thermal-catalytic CO2 methanation reaction. Reprinted from [118], Copyright (2018), with permission from Elsevier. b Schematic showing the plasma-induced catalyst and catalyst-induced plasma synergy. Reprinted with permission from [119]. Copyright (2023) American Chemical Society
The interactions between plasma and catalysts in a DBD reactor are very complex, as illustrated by the proposed mechanisms shown in Fig. 11b. Detailed information about reaction intermediates is required to understand the reaction pathways fully. In turn, this understanding can help design active catalysts for the process. Researchers have been endeavouring to unravel the reaction mechanisms of plasma-assisted methanation using advanced in-situ characterisation tools, including optical emission spectroscopy (OES), X-ray photoelectron spectroscopy (XPS), X-ray absorption spectroscopy (XAS) and diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS). These methods complement each other and help to determine the intermediate species [120, 121]. Computational modelling can assist with determining and validating the reaction pathways. However, because plasma CO2 methanation is an emerging and complex process, limited research has been performed on its mechanisms. In this section, we consider in-situ measurements of surface-adsorbed species (mainly DRIFTS) and gas-phase species (OES), followed by an overview of the reaction kinetic studies that allow the apparent activation energy of the overall reaction to be determined. Computational modelling and simulation are discussed in the following section.
In-situ Measurements of Surface-Adsorbed Species
Plasma-coupled DRIFTS allows in-situ probing of the evolution of absorbed species and intermediates on the catalyst surface. Vibrational frequencies of chemical bonds and functional groups on the catalyst surface can be identified, helping to determine reaction pathways.
Xu et al. [90] used the self-designed plasma DRIFTS–mass-spectroscopy setup shown in Fig. 12a to compare thermal and plasma-assisted CO2 methanation over Ru/MgAl-R300 catalysts. The presence of a plasma changed the observed surface intermediates from CO only to include formyl (HCOad at 1756 and 1132 cm−1), carbonyl (COad at 2023 and 1950 cm−1) and carbon-hydroxyl (COHad at 1300 cm−1) species, where subscript ‘ad’ denotes surface-adsorbed; the spectrum is shown in Fig. 12b. The formyl and carbon hydroxyl species signals increased at a rate consistent with CH4 production, suggesting they were hydrogenated on the Ru surface to produce CH4, as per the mechanism illustrated in Fig. 12c. The mechanism proposed by Xu et al. included both dissociative adsorption of CO2 to form COad and Oad and adsorption of plasma-produced to form COad, before hydrogenation of COad to form HCOad and CHOad.

a Schematic view of in-situ DRIFTS cell designed for plasma-assisted gas phase catalysed species [122]. Reproduced with permission from Springer Nature. b In-situ DRIFTS spectra of surface species on 2.5%Ru/MgAl catalyst under plasma-on condition (5.0 kV, 23.5 kHz; the labelled peaks are identified in the text and the large peaks at 2361 and 2343 cm−1 correspond to gas-phase CO2. Reproduced from [90]. CC BY 4.0. c Mechanism of NTP activated CO2 hydrogenation over 2.5%Ru/MgAl catalysts. Reproduced from [90]. CC BY 4.0. d Temperature-programmed methanation of a feed of 12CO2/H2/Ar on Co/CeZrO4 on which 13CO2 was pre-adsorbed. Reproduced from [123]. CC BY-NC-ND 4.0
Parastaev et al. [123] used a temperature-programmed plasma surface reaction (TPPR) method, combined with isotopically labelled CO2, to decouple the contributions of gas-phase reactions from the plasma-induced surface reactions. The activation temperature for flowing 12CO2 was found to be 50 ℃ lower than for pre-absorbed 13CO2, indicating the contribution of the gas discharge to the low-temperature activation—see Fig. 12d. The authors proposed that CO formed by gas-phase dissociation of CO2 was an important intermediate species and confirmed this using TPPR with pre-adsorbed CO.
Chen et al. [84] compared the methanation reaction mechanisms for plasma-assisted and thermal catalysis over 15Ni-20La/Na-Beta using in-situ DRIFTS. Their findings demonstrated that a temperature higher than 350 °C was required to overcome the reaction energy barrier for thermal methanation, while under plasma conditions, the catalyst was activated at a lower temperature (< 150 °C). Without plasma, the IR bands due to OH vibrational stretching were located at 3500–3800 cm−1 and 1500–1800 cm−1. These OH groups facilitated CO2 adsorption on the catalyst surface. When the plasma was turned on, IR bands of carbonate (at 1653, 1433, 1292 cm−1) and monodentate formate (at 1561 cm−1) were detected, suggesting the formation of formates on the catalyst surface, as shown in Fig. 13a. These surface species were further hydrogenated to CHx species and subsequently formed CH4. In contrast, for thermal reactions, the bands of carbonate and monodentate formate species gradually started appearing only above 150 °C, and their magnitude increased with temperature up to 400 °C, demonstrating a higher temperature was required to overcome the reaction barrier (Fig. 13b). Chen et al. also found the H radicals in the gas phase may react with surface-adsorbed species. Therefore, the plasma-assisted reactions proceeded via both L–H and Eley–Rideal (E-R) mechanisms, while the thermal reactions only proceeded via the L–H mechanism.

In-situ DRIFTS spectra of species generated on 15Ni-20La/Na-Beta during a plasma-assisted and b thermal CO2 methanation. Reproduced from [84]. CC BY 3.0
Chen et al. [75] examined plasma-assisted CO2 methanation over the MOF-based catalyst 15Ni/UiO-66 using in-situ DRIFTS coupled with mass spectrometry. After the plasma was turned on, the band intensities of the gas-phase CO2 (at 2340 cm−1) and the OH bands (at 1500–1800 cm−1) started disappearing while those of surface-adsorbed carbonate and formate species (at 1561, 1433, and 1292 cm−1) progressively increased. These trends are consistent with the results of Chen et al. [84] discussed above. Chen et al. [75] proposed that, under plasma conditions, excited CO2 species were adsorbed on Ni sites and then formed carbonate species after reacting with hydroxyl groups on the catalyst surface. These carbonate species then reacted with either surface-adsorbed or gas-phase H to form formate, which was subsequently hydrogenated to CH4. The authors also compared the NTP catalytic activity of 15Ni/UiO-66 with two reference catalysts 15Ni/α-Al2O3 and 15Ni/ZrO2, finding the MOF-based catalyst showed greatly enhanced CO2 conversion. In-situ DRIFTS spectra of the two reference materials suggested that the plasma had little effect on the gas-phase CO2 or surface species bands, in accordance with the CO2 conversion results. The results suggest the carbonate and formate species formed on the catalyst surface under plasma conditions are the key steps for CH4 formation.
Wang et al. [71] studied the methanation reaction pathways for a Co/Al2O3 catalyst in a DBD reactor. An analysis of reaction kinetics and in-situ DRIFTS, including experiments using pre-adsorbed H2 or CO2, demonstrated that H radicals were formed via collisions with highly energetic electrons in the plasma, that their adsorption on the catalyst surface was vital for the methanation reaction and that methane was not generated by E-R reactions of gas-phase H with surface-adsorbed carbon species. However, fewer surface-adsorbed hydrogen species were observed for plasma-assisted than thermal methanation. The authors proposed that while the L–H pathway is likely to be important, different reaction pathways also existed. In particular, plasma-activated CO2 and/or CO formed in the plasma reacted with surface-adsorbed H on the metallic Co surface through the E-R mechanism and subsequently hydrogenated to CH4.
Azzolina-Jury et al. [124] investigated the mechanisms of the low-pressure plasma-assisted methanation reaction using operando time-resolved FTIR spectroscopy over an Ni/USY catalyst. They observed that gas-phase excited CO2 and CO were adsorbed on Ni sites as monodentate formates and carbonyls, respectively. The formates were subsequently hydrogenated to linear carbonyls. The carbonyls were then hydrogenated to CH, CH2, CH3 and CH4 molecules. The low CH4 selectivity observed was believed to be due to the low adsorption of CO2 on the zeolite support.
In summary, in-situ plasma DRIFTS analysis has proven very useful for reaction pathway investigation. According to the research outcomes reported in the literature, there are two major roles that plasma plays in the process: (a) facilitate the excitation and dissociation of CO2 by electron impact reactions; (b) modulate the surface reaction pathway by producing relevant intermediates at the interface. Production of H radicals may also be important. The relative importance of gas-phase CO2 and CO is strongly debated.
It is worth mentioning this method is still in an early stage of development. It is challenging to differentiate the IR bands and other signals originating from structural bands and photons and correlate them with structural changes and intermediate components. The proposed mechanisms for different catalyst systems vary with the active metal and support materials used. Questions about the role of the active metal and its interaction with the plasma and active species remain largely unanswered.
In-situ Measurements of Gas-Phase Species: Optical Emission Spectroscopy
OES is an important plasma diagnostic technique that enables the measurement of in-situ plasma reactive species (excited atoms, ions and molecules) in the gas phase and in some cases, rotational and vibrational temperatures of molecules. The measurements can provide detailed information about reaction intermediates, helping to understand the reaction mechanisms. Considering the importance of ladder-climbing effects and the Eley–Rideal mechanism in plasma-assisted CO2 activation, information about the vibrational excitation distribution is of particular interest. As shown in Fig. 14a and b, the specific spectra lines of excited CO, CH, H2 and H originating from the H2/CO2 activation and relevant radical recombination reactions have been measured [113]. The vibrational–rotational bands of CO (b3Σ → a3Π) and CO (B1Σ → A1Π) showed that CO2 molecules were activated into vibrationally excited CO species. The bands of CH (A2△ → X2Π, 0–0), CH (B2Σ → X2Π, 0–0) and H2 Fulcher and the Hα line indicated the production of vibrationally excited H2 species, H atoms and CH4 fragments (CH). Similar observations were obtained by Mikhail and Guo et al. [81, 85]. Gao et al. [82] found that the electron-induced reactions were highly dependent on the operating parameters (e.g., applied voltage, repetition frequency, discharge gap length) by using a highly adjustable nanosecond pulse power. The Boltzmann equation solver BOLSIG + [125] and cross-section data from the LXCAT database were applied to determine the relationship between the reduced electric field and the electron energy loss fractions for electron-induced excitation reactions. Additionally, owing to the important role of the COx vibrational excitation states, high-resolution OES and the Treanor equation [126] were used to examine the vibrational distribution functions of CO for specific vibrational temperatures, as shown in Fig. 14c. About 60% of the CO molecules were in the ground state, and 98% were at or below the fourth vibrationally excited state.

a Emission spectra of H2/CO2 DBD for different reactors. Reprinted with permission from [113]. Copyright (2018) American Chemical Society. b Comparison of typical optical emission spectra for plasma-only and plasma-catalytic methanation [82]. c Plasma-induced vibrational excitations of CO for Boltzmann and Treanor vibrational distribution functions of CO at Tvib = 3500 K and Tgas = 510 K. Reprinted with permission from [82], Copyright (2021), with permission from Elsevier
Though OES can be a very reliable diagnostic technique to detect gas-phase species, it uses an optical fibre to collect emitted light. Anything that interferes with the light path, such as dielectric materials, electrodes and other species coming from the discharge, can affect the results. Therefore, caution is required when interpreting the OES spectral data.
Reaction Kinetics Studies
Kinetic studies are important to determine the kinetic parameters such as rate constant, activation energy and reaction order. Kinetic studies of plasma catalysis also allow us to determine the effect of various input variables, such as discharge power, concentration of reactants, and feed ratio, on the performance and efficiency of the process.
Mu et al. [127] calculated the activation energy for thermal and plasma-assisted CO2 methanation using Ni/SiO2 catalysts using Eqs. (13) and (14), respectively.
where TOF is the turnover frequency, Ethermal and Eplasma are the thermal and plasma-assisted activation energies, respectively, PDBD is the DPD plasma power, and Ftotal is the total flow rate of the feed gas mixture. CO2 conversion in thermal catalysis displayed Arrhenius behaviour, as demonstrated by the Arrhenius plot shown in Fig. 15a, with Ethermal = 80 \(\pm\) 3 kJ/mol. In contrast, for plasma-assisted catalysis, the temperature dependence of the turnover frequency was found to be non-exponential, thus exhibiting non-Arrhenius behaviour. Kim et al. [128] obtained a linear correlation between ln(TOF) and the reciprocal of the DBD plasma power (1/PDBD) instead of 1/T, as demonstrated in Fig. 15b. The DBD plasma power controlled the density of electrons in the gas phase, which directly affected the rate of excited species (CO2* and H*). Meanwhile, they also suggested that plasma-induced vibrational excitation of species, such as CO2*, which reacted with dissociated H* species in the presence of the Ni catalyst, lowered the activation barrier to 29 kJ/mol, almost 63% lower than that of thermal CO2 hydrogenation.

a Arrhenius plot of thermal-catalytic CO2 hydrogenation over Ni/SiO2 catalyst; b plot of ln (TOF) as a function of the reciprocal of the DBD power and linear fits for plasma-assisted catalytic CO2 hydrogenation over Ni/SiO2 catalyst. Reprinted with permission from [127]. Copyright (2020) American Chemical Society
The activation energy barrier for plasma-assisted CO2 methanation reaction over Ru catalyst loaded on SiO2 and Al2O3 supports was investigated by Xu et al. [72]. They showed the activation energy for thermal CO2 methanation on Ru/SiO2 was 66.2 kJ/mol, but was reduced to 20 kJ/mol for the plasma-assisted process, explaining why the thermal process required high temperatures, 260–320 °C, but the plasma-assisted process proceeded at lower temperatures (< 129 °C). The kinetic studies, coupled with in-situ DRIFTS, further indicated that plasma-assisted catalysis involved both L–H and E-R mechanisms.
Xu et al. also investigated the effect of CO poisoning on the surface of catalyst for thermal and plasma conditions. Their findings revealed that in thermal catalysis, CO poisoning can deactivate the catalyst as CO is preferentially adsorbed on the Ru surface, inhibiting the adsorption of CO2 and H2. For plasma-assisted CO2 methanation, the plasma excited CO2 molecules, facilitating the adsorption of CO2. In addition, more active sites for CO2 adsorption were available as collisions with plasma species removed the strongly adsorbed CO, leaving the sites available for other reactive species. As a consequence, CO poisoning was reduced to a benign level under plasma conditions; the plasma could both mitigate the occurrence of CO poisoning and regenerate previously poisoned catalysts.
Wang et al. [71] performed a comparative study of CO2 hydrogenation under thermal and plasma-assisted catalytic conditions using an alumina-supported cobalt catalyst. Significant plasma-catalyst synergy was revealed in the low-temperature range between 423 and 498 K, where the CO2 conversion was 44%, which was 3.6 times the sum of conversion achieved by thermal catalysis and the plasma-alone process. The plasma-catalytic reaction was found to have an apparent activation energy (∼40 kJ/mol) less than half of that in the thermal catalytic reaction (∼87 kJ/mol). The authors proposed that the promotion effect was likely due to a new RWGS reaction pathway introduced by plasma, with the CO formed in the plasma reaction with plasma-induced H species adsorbed on the cobalt surface, as explained above in the “In-situ Measurements of Surface-Adsorbed Species” sub-section.
Chen et al. [75] also studied the kinetics of the CO2 methanation reaction under thermal and plasma-assisted catalytic conditions. They reported that the presence of the plasma reduced the activation energy by 55% (from 70 to 32 kJ/mol). Under thermal conditions, the reactants (CO2 and H2) were first adsorbed on the catalyst surface. Subsequently, the surface-dissociated H species reacted with surface-adsorbed CO2 species via the L–H mechanism. Mu et al. [127] reported similar surface adsorption reactions for thermal catalysis. The input heat energy (temperature) was the main factor controlling the dissociation of molecules on the surface to promote surface reactions. In contrast, the plasma promoted the vibrational and electronic excitation of CO2 and dissociation of H2, which allowed them to interact with catalyst surface and resulted in lower activation energy barrier, which was confirmed by in-situ analysis.
Gao et al. [82] analysed the kinetics of plasma-assisted and thermal catalytic methanation reactions using bimetallic Ni–Fe catalysts. The dissociative adsorption of CO on the catalyst surface was considered to be the rate-determining step in the thermal process, with the highest energy barrier of 237.4 kJ/mol. The activation barrier can be lowered with the presence of vibrationally excited species near interfacial active sites. Vibrationally excited CO molecules were observed in the plasma-assisted case, lowering the apparent activation barrier to 36 kJ/mol compared to 82 kJ/mol for thermal catalysis.
The studies all demonstrated that coupling plasma with catalytic materials generated vibrationally excited species in the gas phase, providing alternative pathways that lowered the activation barrier compared to their counterpart thermal reactions and promoted methanation at much milder temperatures.
Modelling and Simulation
Experimental investigations are very unlikely to provide a full understanding of the reaction mechanisms in a complex process such as plasma catalysis. As a consequence, computational modelling and simulation have assumed an important complementary role. They can provide fundamental information about reaction mechanisms, determine the relative importance of different reaction paths, and provide insights into physical processes, including heat transfer, flow and plasma–surface interactions.
Extensive modelling studies have been performed to gain a fundamental understanding of the plasma and surface chemistry in plasma catalysis [129], particularly in fields of CO2 hydrogenation and N2 reduction. The results have demonstrated that many underlying mechanisms of plasma catalytic reactions can be explained by different modelling techniques [130, 131]. In this section, we review the modelling and simulation of CO2 hydrogenation under non-thermal plasma conditions, focusing on the CO2 methanation process.
Density Functional Theory (DFT) Calculations
DFT calculations can be used to describe semi-quantitatively the kinetics of CO2 activation on the basis of thermochemistry and activation energies [132,133,134]. Three different mechanisms of thermal catalytic CO2 methanation at a molecular level have been proposed. Ren et al. [135] calculated the relative energy changes of the three reaction pathways over an Ni(111) surface. The first pathway (path 1) involves the reaction between CO2 and H to produce HCOO species, which then dissociate into CO and OH, followed by the hydrogenation of CO to CH4. The rate-determining step in this pathway is the conversion of HCOO to CO + OH, with an energy barrier of 306.8 kJ/mol. The second pathway (path 2) involves the decomposition of CO2 into CO and O on Ni(111), followed by the dissociation of CO into C and O, and then the stepwise hydrogenation of C to CH4. The rate-determining step is the elementary reaction CO → C + O, with an energy barrier of 237.4 kJ/mol. In the third pathway (path 3), CO2 reacts with H to produce C(OH)2 species, which then dissociate into CH2O and OH species, and CH2O further dissociates into CH2 species. The rate-determining step in this pathway is the formation of C(OH)2 on the Ni(111) surface, with an energy barrier of 292.3 kJ/mol. The potential energy diagram of the three processes is shown in Fig. 16. According to the calculated energy barrier of each reaction step, path 2 is the optimal mechanism.

Potential energy diagram of three mechanisms of the thermal CO2 methanation process on an Ni(111) surface. Reprinted from [135], Copyright (2015), with permission from Elsevier
The above DFT calculations provide a basic understanding of the CO2 methanation process, assuming direct CO2 activation occurs only over the catalyst surface. However, plasma catalysis involves excited species and radicals.
Only a few studies have employed DFT simulations to investigate plasma-enabled CO2 methanation. Experimental studies have found that HCOO is an important intermediate species [73, 75, 136]. Kim et al. [136] focused on the HCOO formation as the most favourable pathway during CO2 hydrogenation, using DFT simulations of the thermal catalytic system combined with measurements of surface-adsorbed species (discussed in the “In-situ Measurements of Surface-Adsorbed Species” sub-section) to draw conclusions about the mechanisms in the presence of a plasma. Their calculated energy diagram and the corresponding structures are shown in Fig. 17a and b. E-R and L–H pathways were considered for the initial activation of CO2(g) to monodentate HCOO formation on the Pd2Ga (020) surface, with the E-R pathway (reaction of CO2(g) with surface-adsorbed H) found to be favoured even under thermal conditions. The apparent activation energy of the DBD reactions is estimated to be ca. 43 kJ/mol, which is smaller than the activation barrier for monodentate HCOO formation (67.7 kJ/mol) and decomposition (74.1 kJ/mol); not only HCOO formation via the E-R pathway but also the undesirable decomposition of HCOO to CO is promoted by DBD. In accordance with the measurements of surface-adsorbed species, the DFT calculations showed the decomposition of HCOO to yield CO occurred via the spillover of H atoms adsorbed on metallic particles. The role of DBD is, therefore, not limited to the vibrational excitation of CO2, but the activation of H2, leading to enhanced hydrogen spillover.

DFT calculations of the CO2 hydrogenation reactions on Pd2Ga (020): a energy diagram (m-HCOO and b-HCOO denote monodentate and bidentate formate, respectively); b corresponding DFT optimised geometries, showing the energy barrier (colour scheme: black: Pa, green: Ga, grey: C, red: O, white: H). Reprinted with permission from [136]. Copyright (2022) American Chemical Society. c Reaction pathways of CO2 hydrogenation to CH3OH and CH4; species adsorbed at the active sites are labelled as “*”; to make the figure more readable, H2 decomposition and H2O desorption are omitted from the pathways. Reprinted with permission from [137]. Copyright (2022) American Chemical Society (Color figure online)
Recent studies have shown that the type of heterogeneous catalyst used is crucial in influencing the product distribution, particularly with regards to C1 selectivity (CO, CH4, CH3OH). Yi et al. [137] demonstrated a synergistic effect between a Cu/γ-Al2O3 catalyst and the CO2/H2 plasma, achieving a CO2 conversion of 10% at 4 wt% Cu loading and a CH3OH selectivity near 50%. DFT calculations showed that the path to CH4 formation begins with the decomposition of H3CO*, as shown in Fig. 17c. The cleavage of the C-O bond for H3CO* species needed to overcome a high barrier (2.03 eV), much higher than in H3CO* hydrogenation. Although the hydrogenation of CH3* to CH4 (g) displayed a low barrier of 0.66 eV, the following hydrogenation steps for the decomposition of O* to OH* and further to H2O* also showed high barriers (1.87 and 1.64 eV). Therefore, the DFT results indicated that CH4 formation via the above path has much higher barriers than CH3OH production, which can explain the much lower yield of CH4 in the experiment.
DFT simulation is of particular relevance to examining the importance of possible intermediate species. Combining DFT with DRIFTS measurements provides a valuable tool for studying and validating reaction pathways. The approach is particularly appealing for plasma catalysis because of its complexity, and such studies are starting to become more common. Nevertheless, very few DFT simulations related to plasma CO2 methanation have been performed, and further work will be of great value.
Chemical Kinetic and Fluid Modelling
Zero-dimensional chemical kinetic modelling is widely applied to plasma processes, including those that incorporate catalysts. They are based on solving continuity equations for species in the plasma or on surfaces. A Boltzmann equation solver is usually incorporated to calculate the electron energy distribution function, on which the rate coefficients for electron impact reactions depend. Bogaerts et al. [54] provided a good summary of this and other modelling approaches.
A zero-dimensional plasma kinetics model was developed by Du et al. [37] to investigate the reaction pathways of plasma-assisted catalytic CO2 hydrogenation. The continuity equations were solved assuming spatially homogenous discharge conditions. Results were obtained for the production of CH4 on Ni(111) and CH3OH on Cu(111). For Ni(111), CH4 formation mainly occurs by successive E-R reactions of C(s) with gas-phase H to form CH(s), CH2(s), CH3(s) and then eventually CH4, as shown in Fig. 18a. Although H(s) is the dominant surface-adsorbed species, E-R and L–H reactions involving H(s) were less important. The mechanism by which C(s) is produced was not discussed, although the density of C(s) was 100 times higher than that of CO(s). L–H reactions promoted the formation of the intermediate species CHx(s), which indirectly promoted the CH4 formation. The rates of the reactions to generate CHx(s) and CH4 are in the range of 1010–1018 cm−3s−1, as shown in Fig. 18b, with the corresponding reactions listed in Table 4.

a Mechanism of CO2 hydrogenation using Ni (111) catalyst; b reaction rates related to CHx(s) species and CH4 [37]. © 2022 Wiley-VCH GmbH
Gao et al. [82] and Chen et al. [138] also developed a zero-dimensional kinetic model to study plasma-assisted CO2 hydrogenation, but only gas-phase reactions were considered. Gao et al. predicted the production of high densities of vibrationally excited CO, which they proposed to be an important precursor in the catalytic production of CH4. Electron impact dissociation of CO2 was the main mechanism for CO formation. Chen et al. [138] examined the influence of the CO2/H2 ratio in the feed gas on the production of CO, CH4 and CH3OH.
Microkinetic models, which consider only reactions involving at least one surface-adsorbed species, are widely used in thermal catalysis studies. Plasma effects can be included by considering atoms, radical and excited molecules. Michiels et al. [139] developed a mean-field microkinetic model for CO2 hydrogenation to CH3OH on a Cu(1,1,1) surface, using the fluid model of De Bie et al. [40] to provide the plasma composition. An interesting finding, possibly relevant to CO2 methanation, was that vibrational excitation of CO2 increases the CH3OH production rate.
Surface reactions on suitable catalysts are critical in the production of CH4, so studies that do not consider such reactions are of limited value in understanding the mechanisms of CO2 methanation. While Du et al. [37] did consider reactions on an Ni(111) surface, they did not include any involving CHxOy species (although such species were considered for CH3OH production on the Cu(111) surface). As discussed in the “In-situ Measurements of Surface-Adsorbed Species” and “Density Functional Theory (DFT) Calculations” sub-sections, measurements of surface-adsorbed intermediate species and DFT calculations point to the importance of CHxOy species. There is clear scope for additional chemical kinetic modelling, including microkinetic modelling, to assist in understanding the reaction mechanism.
1D and 2D models allow the transport of species to be considered in addition to their chemical reactions. The application of such models in plasma catalysis has been limited to studies of gas-phase reactions and investigations of the interactions of plasma with pores [54]. Here we briefly mention relevant gas-phase models. De Bie et al. [40] investigated the formation of different hydrocarbons by CO2 hydrogenation using a one-dimensional fluid model MD2D, part of the Plasimo package. The model was applied to a cylindrical plasma reactor, assuming a homogenous plasma. The species included in the model were taken from previous calculations for similar systems, including CH4/O2 and CH4/CO2 gas mixtures [140]. The species continuity, drift–diffusion and electron energy balance equations were coupled to the Poisson equation for electric field calculation. The predicted reaction products were mainly CO, CH4 and H2O, with some CH2O, C2H6 and CH3OH also present.
Liao et al. [141] developed a two-dimensional fluid model to investigate CO2 hydrogenation in a DBD reactor, including the spatial density distribution of ions, radicals, and streamer propagation distribution and their influence.
Fluid models can be a valuable source of provide species concentration data for use in microkinetic models; as noted above, the 1D results of De Bie et al. [40] were used in a microkinetic model of CO2 hydrogenation to form CH3OH and could equally be applied to modelling CH4 production.
Conclusions and Future Research Needs
We have reviewed the current status of plasma-assisted CO2 methanation, including catalysts, reaction mechanisms, diagnostics and modelling, and the influence of process parameters control and reactor settings. Plasma-assisted CO2 methanation is an emerging area with immense promise. High CO2 conversion and CH4 selectivity have already been demonstrated at low temperatures. The best fuel production efficiencies obtained are competitive with the thermal process that is currently used for industrial CO2 methanation. Moreover, DBD reactors are suited to the intermittent nature of renewable energy, can run at ambient conditions and are easily scalable for industrial applications. Nevertheless, it is important to underline that plasma catalytic CO2 methanation is still at an early stage of development. The process needs to be investigated in greater detail to understand the complex physics and chemistry involved. The following directions are of particular relevance.
Catalyst Materials Development
Of the limited range of catalysts explored for plasma-assisted CO2 methanation, most are Ni-based. However, stability and catalyst lifetime haven’t been examined thoroughly in plasma conditions. Other metals such as Co, Rh, Ru and Pd were also used, some of which demonstrated good performance, but none outperformed Ni. Bimetallic catalysts have not been assessed in any depth. A range of support materials, including metal oxides, MOFs and zeolites, has been tested. The choice of support affects catalyst performance by altering properties such as the dielectric constant, porosity and affinity for the water product, the influence of all of which requires further investigation. The use of CeO2 as a promoter in support materials has been shown to enhance CO2 conversion and CH4 selectivity, possibly because it improved the Ni dispersion and the distribution of basic sites or increased the dielectric constant. Many materials that could be used as support materials to facilitate the active metal’s function remained unexamined.
Surface Basicity
Surface basicity is an important factor in CO2 methanation. In particular, the medium-strength basic sites are considered responsible for the formation of monodentate formate species, which are seen as important intermediates for methane formation. Different active metals and supports offer different strengths of basic sites for CO2 adsorption. The role of the basicity of the support under plasma conditions and means for manipulating the basicity remain largely unexplored.
Catalyst Bed Temperature
The real-time measurement of the temperature of the catalyst bed in a DBD plasma is very challenging due to electromagnetic interference and plasma luminescence [142, 143]. Therefore, most of the reported temperatures for plasma catalytic reactions are based on outer wall temperatures of the reactor [80] or temperatures measured by thermocouples after the plasma is turned off or located at the exit of the gas outlet [80, 120, 142]. Heat transfer modelling [144] and measurements using a shielded probe inserted in the high-voltage electrode [145] indicate that the wall temperature is significantly lower than the catalyst bed temperature, although by significantly different amounts. Even if the temperature is known, its influence on the plasma methanation process is difficult to determine since the temperature is a function of the applied voltage and gas flow rate, which also influence the process [144, 145]. While it has been demonstrated that plasma-assisted CO2 methanation occurs at catalyst bed temperatures as low as 116 °C [144], the influence of temperature on the discharge behaviour of the DBD reactor and its synergistic impact on the CO2 methanation remain poorly understood and constitute an important area for further investigation.
Physical Properties of the Catalysts
Plasma-assisted CO2 methanation is mostly in packed-bed DBD plasma reactors. Modelling has revealed that the electric field is typically enhanced at the contact points between the beads, increasing electron impact excitation, ionization, and dissociation. Some early modelling research suggested that the electron impact reactions are closely related to the materials’ dielectric constant and porosity. Smaller pore sizes only led to enhanced ionisation for lower dielectric constants, i.e., up to εr = 200, 150, and 50 for pore sizes of 50, 30, and 10 μm, respectively. Ferroelectric materials with dielectric constants above 300 were not associated with increased ionisation, even for 100 μm pore sizes [146]. In recent years, many novel synthesis methodologies have been developed to tailor the structure and porosity of the materials. However, experimental studies of the influence of pore sizes and dielectric constants for plasma CO2 methanation are very limited. It is valuable to conduct more research in this area to generate an understanding of how these physical properties affect the plasma process and how to tailor the materials for plasma catalysis.
Catalyst Pre-treatment
Catalyst pre-treatment has gained significant attention because plasma consists of highly excited species that can change the catalyst’s chemical and physical properties. Through etching, doping, sputtering, and heating, plasma creates new surface properties, such as oxygen vacancies, surface functional groups and surface defects. However, the optimum frequency and duration of pre-treatment and the dependence of these parameters on the type of catalyst have not been thoroughly studied.
Reaction Mechanisms
A detailed understanding of plasma catalysis is instrumental in improving the process, developing better catalysts and increasing fuel production efficiency. Unlike thermal catalysis, which proceeds mainly via L–H interactions on the catalyst surface, NTP produces gas-phase excited species, which can react with surface-adsorbed species on the catalysts via E-R as well as L–H mechanisms, thus reducing the activation barrier. These hybrid plasma catalytic systems are complex and require advanced characterisation to fully understand the phenomena occurring in the gas phase and on the surface. Various advanced in-situ characterisation tools such as DRIFTS, XAS, XPS and OES are available to detect the intermediate species and catalyst surface chemical states to help understand the active species and their interaction with the catalyst surface. By doing so, reaction pathways involved in the plasma methanation reaction can be revealed. The parallel application of modelling methods such as DFT, kinetic modelling and fluid modelling can be powerful in validating hypotheses by providing insights into surface and gas-phase reactions, and the densities of excited species, radicals, and surface-adsorbed species.
Process Optimisation
The performance in plasma-assisted CO2 methanation is strongly dependent on the range of operational parameters. Parameters such as excitation voltage and frequency, diluent gas, and reactor packing material are found to affect the properties of the discharge and, so, the performance of the reactor. However, assessing the importance of each of the parameters is difficult since they are not independent; for example, the packing material affects the reaction kinetics by altering electric field distribution and surface reactions. The choice of optimum process parameters remains a significant challenge and requires systematic studies, ideally including in-situ diagnostics and modelling.
Reactor Design
A few different types of DBD reactors have been studied for CO2 methanation. The reactors are all based on simple designs with a high-voltage electrode on the axis and a mesh, foil or water jacket as the ground electrode on the outside. The reduced electric field in a DBD is typically over 100 Td, which favours dissociation over vibrational excitation of molecules, reducing the energy efficiency of the process. Studies on optimising the reactor design to promote the desired gas-phase reactions and recycling the waste heat from the process to increase energy efficiency are limited. Even though plasma processes are well suited for the scale and intermittent nature of renewable energy sources, improved reactor design to improve efficiency is critical for their large-scale commercialisation.
Plasma-assisted CO2 methanation has the potential to be an important industrial technology. There are no obvious fundamental barriers to the further development and ultimate implementation of the technology. Research addressing the points itemised above will lead to the optimised catalysts, process parameters and reactor design necessary to improve the conversion, selectivity and fuel production efficiency further, pointing the way to industrial implementation.
Data Availability
Not applicable.
Abbreviations
- NTP:
-
Non-thermal plasma
- DBD:
-
Dielectric barrier discharge
- GA:
-
Gliding arc
- MW:
-
Microwave
- MOF:
-
Metal organic framework
- RWGS:
-
Reverse water gas shift
- L-H:
-
Langmuir-Hinshelwood
- DRIFTS:
-
Diffuse reflectance infrared Fourier transform spectroscopy
- SEI:
-
Specific energy input
- OES:
-
Optical emission spectroscopy
- LHV:
-
Lower heating value
- RF:
-
Radio frequency
- E-R:
-
Eley-Rideal
- EC:
-
Energy cost
- FPE:
-
Fuel production efficiency
- LDH:
-
Layered double hydroxide
- FTIR:
-
Fourier transform infrared
References
Gao J, Liu Q, Gu F, Liu B, Zhong Z, Su F (2015) Recent advances in methanation catalysts for the production of synthetic natural gas. RSC Adv 5(29):22759–22776. https://doi.org/10.1039/C4RA16114A
Wang W, Wang S, Ma X, Gong J (2011) Recent advances in catalytic hydrogenation of carbon dioxide. Chem Soc Rev 40(7):3703–3727. https://doi.org/10.1039/C1CS15008A
Lee WJ, Li C, Prajitno H, Yoo J, Patel J, Yang Y, Lim S (2021) Recent trend in thermal catalytic low temperature CO2 methanation: a critical review. Catal Today 368:2–19. https://doi.org/10.1016/j.cattod.2020.02.017
Miguel CV, Mendes A, Madeira LM (2018) Intrinsic kinetics of CO2 methanation over an industrial nickel-based catalyst. J CO2 Util 25:128–136. https://doi.org/10.1016/j.jcou.2018.03.011
Falbo L, Martinelli M, Visconti CG, Lietti L, Bassano C, Deiana P (2018) Kinetics of CO2 methanation on a Ru-based catalyst at process conditions relevant for Power-to-Gas applications. Appl Catal B 225:354–363. https://doi.org/10.1016/j.apcatb.2017.11.066
Ronsch S, Kochermann J, Schneider J, Matthischke S (2016) Global reaction kinetics of CO and CO2 methanation for dynamic process modeling. Chem Eng Technol 39(2):208–218. https://doi.org/10.1002/ceat.201500327
Gallandat N, Mutschler R, Vernay V, Yang H, Zuttel A (2018) Experimental performance investigation of a 2 kW methanation reactor. Sustain Energ Fuels 2(5):1101–1110. https://doi.org/10.1039/c8se00073e
Aziz MAA, Jalil AA, Triwahyono S, Mukti RR, Taufiq-Yap YH, Sazegar MR (2014) Highly active Ni-promoted mesostructured silica nanoparticles for CO2 methanation. Appl Catal B 147:359–368. https://doi.org/10.1016/j.apcatb.2013.09.015
Zhang S, Gao Y, Sun H, Bai H, Wang R, Shao T (2018) Time-resolved characteristics and chemical kinetics of non-oxidative methane conversion in repetitively pulsed dielectric barrier discharge plasmas. J Phys D Appl Phys 51(27):274005. https://doi.org/10.1088/1361-6463/aac5ad
Liu Y, Zhang S, Huang B, Dai D, Murphy AB, Shao T (2021) Temporal evolution of electron energy distribution function and its correlation with hydrogen radical generation in atmospheric-pressure methane needle–plane discharge plasmas. J Phys D Appl Phys 54(9):095202. https://doi.org/10.1088/1361-6463/abca61
Dębek R, Azzolina-Jury F, Travert A, Maugé F (2019) A review on plasma-catalytic methanation of carbon dioxide – Looking for an efficient catalyst. Renew Sustain Energy Rev 116:109427. https://doi.org/10.1016/j.rser.2019.109427
Gao Y, Zhang S, Sun H, Wang R, Tu X, Shao T (2018) Highly efficient conversion of methane using microsecond and nanosecond pulsed spark discharges. Appl Energy 226:534–545. https://doi.org/10.1016/j.apenergy.2018.06.006
Snoeckx R, Bogaerts A (2017) Plasma technology – a novel solution for CO2 conversion? Chem Soc Rev 46(19):5805–5863. https://doi.org/10.1039/C6CS00066E
Zeng Y, Tu X (2016) Plasma-Catalytic CO2 hydrogenation at low temperatures. IEEE Trans Plasma Sci 44(4):405–411. https://doi.org/10.1109/TPS.2015.2504549
Neyts EC, Ostrikov K, Sunkara MK, Bogaerts A (2015) Plasma catalysis: synergistic effects at the nanoscale. Chem Rev 115(24):13408–13446. https://doi.org/10.1021/acs.chemrev.5b00362
Gao Y, Dou L, Feng B, Zhang C, Shao T (2023) Catalyst-free activation of CH4 and air into platform chemicals and H2 using parametrized nanosecond pulsed plasma. Energy Convers Manage 276:116570. https://doi.org/10.1016/j.enconman.2022.116570
Liu Y, Dou L, Sun H, Zhang C, Murphy AB, Shao T (2023) Selective clipping of a lignin-derived monomer by plasma for bio-oil upgrading. ACS Sustain Chem Eng 11(1):101–112. https://doi.org/10.1021/acssuschemeng.2c04328
Liu Y, Dou L, Zhou R, Sun H, Fan Z, Zhang C, Ostrikov KK, Shao T (2021) Liquid-phase methane bubble plasma discharge for heavy oil processing: insights into free radicals-induced hydrogenation. Energy Convers Manage 250:114896. https://doi.org/10.1016/j.enconman.2021.114896
Wang X, Gao Y, Zhang S, Sun H, Li J, Shao T (2019) Nanosecond pulsed plasma assisted dry reforming of CH4: the effect of plasma operating parameters. Appl Energy 243:132–144. https://doi.org/10.1016/j.apenergy.2019.03.193
Chen X, Zhang S, Li S, Zhang C, Pan J, Murphy AB, Shao T (2021) Temperature-independent, nonoxidative methane conversion in nanosecond repetitively pulsed DBD plasma. Sustain Energy Fuels 5(3):787–800. https://doi.org/10.1039/D0SE01593H
Wang Z, Zhang Y, Neyts EC, Cao X, Zhang X, Jang BWL, Liu C-j (2018) Catalyst preparation with plasmas: how does it work? ACS Catal 8(3):2093–2110. https://doi.org/10.1021/acscatal.7b03723
Bogaerts A, Centi G (2020) Plasma technology for CO2 conversion: a personal perspective on prospects and gaps. Front Energy Res. https://doi.org/10.3389/fenrg.2020.00111
Adamovich I, Baalrud SD, Bogaerts A, Bruggeman PJ, Cappelli M, Colombo V, Czarnetzki U, Ebert U, Eden JG, Favia P, Graves DB, Hamaguchi S, Hieftje G, Hori M, Kaganovich ID, Kortshagen U, Kushner MJ, Mason NJ, Mazouffre S, Thagard SM, Metelmann HR, Mizuno A, Moreau E, Murphy AB, Niemira BA, Oehrlein GS, Petrovic ZL, Pitchford LC, Pu YK, Rauf S, Sakai O, Samukawa S, Starikovskaia S, Tennyson J, Terashima K, Turner MM, van de Sanden MCM, Vardelle A (2017) The 2017 plasma roadmap: low temperature plasma science and technology. J Phys D Appl Phys 50(32):323001. https://doi.org/10.1088/1361-6463/aa76f5
Ashford B, Tu X (2017) Non-thermal plasma technology for the conversion of CO2. Curr Opin Green Sustain Chem 3:45–49. https://doi.org/10.1016/j.cogsc.2016.12.001
Bogaerts A, Neyts EC (2018) Plasma technology: an emerging technology for energy storage. ACS Energy Lett 3(4):1013–1027. https://doi.org/10.1021/acsenergylett.8b00184
Whitehead JC (2016) Plasma–catalysis: the known knowns, the known unknowns and the unknown unknowns. J Phys D Appl Phys 49(24):243001. https://doi.org/10.1088/0022-3727/49/24/243001
Li K, Liu J-L, Li X-S, Zhu X, Zhu A-M (2016) Warm plasma catalytic reforming of biogas in a heat-insulated reactor: dramatic energy efficiency and catalyst auto-reduction. Chem Eng J 288:671–679. https://doi.org/10.1016/j.cej.2015.12.036
Sun SR, Wang HX, Mei DH, Tu X, Bogaerts A (2017) CO2 conversion in a gliding arc plasma: performance improvement based on chemical reaction modeling. J CO2 Util 17:220–234. https://doi.org/10.1016/j.jcou.2016.12.009
Li J, Dou L, Gao Y, Hei X, Yu F, Shao T (2021) Revealing the active sites of the structured Ni-based catalysts for one-step CO2/CH4 conversion into oxygenates by plasma-catalysis. J CO2 Util 52:101675. https://doi.org/10.1016/j.jcou.2021.101675
Bogaerts A, Kozák T, van Laer K, Snoeckx R (2015) Plasma-based conversion of CO2: current status and future challenges. Faraday Discuss 183:217–232. https://doi.org/10.1039/C5FD00053J
de la Fuente JF, Moreno SH, Stankiewicz AI, Stefanidis GD (2016) Reduction of CO2 with hydrogen in a non-equilibrium microwave plasma reactor. Int J Hydrogen Energy 41(46):21067–21077. https://doi.org/10.1016/j.ijhydene.2016.08.032
Li J, Dou L, Liu Y, Gao Y, Hu X, Yu F, Li J, Zhang S, Shao T (2023) One-step plasma reforming of CO2CH4 into hydrogen and liquid fuels: the roles of Cu and Fe sites on products distribution. Fuel Process Technol 242:107648. https://doi.org/10.1016/j.fuproc.2022.107648
Li J, Gao Y, Dou L, Zhang C, Du J, Shao T (2023) Parametric investigations on plasma-activated conversion of CH4–CH3OH to C2–C4 alcohols by nanosecond pulsed discharge. Plasma Process Polym 20(2):2200162. https://doi.org/10.1002/ppap.202200162
Zhang S, Zong L, Zeng X, Zhou R, Liu Y, Zhang C, Pan J, Cullen PJ, Ostrikov K, Shao T (2022) Sustainable nitrogen fixation with nanosecond pulsed spark discharges: insights into free-radical-chain reactions. Green Chem 24(4):1534–1544. https://doi.org/10.1039/D1GC03859A
Zeng X, Zhang S, Liu Y, Hu X, Ostrikov KK, Shao T (2023) Energy-efficient pathways for pulsed-plasma-activated sustainable ammonia synthesis. ACS Sustain Chem Eng 11(3):1110–1120. https://doi.org/10.1021/acssuschemeng.2c06259
Zhang S, Gao Y, Sun H, Fan Z, Shao T (2022) Dry reforming of methane by microsecond pulsed dielectric barrier discharge plasma: optimizing the reactor structures. High Volt 7(4):718–729. https://doi.org/10.1049/hve2.12201
Du J, Zong L, Zhang S, Gao Y, Dou L, Pan J, Shao T (2022) Numerical investigation on the heterogeneous pulsed dielectric barrier discharge plasma catalysis for CO2 hydrogenation at atmospheric pressure: effects of Ni and Cu catalysts on the selectivity conversions to CH4 and CH3OH. Plasma Process Polym 19(2):1–14. https://doi.org/10.1002/ppap.202100111
Snoeckx R, Bogaerts A (2017) Plasma technology-a novel solution for CO2 conversion? Chem Soc Rev 46(19):5805–5863. https://doi.org/10.1039/c6cs00066e
Xu S, Chen H, Hardacre C, Fan X (2021) Non-thermal plasma catalysis for CO2 conversion and catalyst design for the process. J Phys D Appl Phys 54(23):233001. https://doi.org/10.1088/1361-6463/abe9e1
De Bie C, van Dijk J, Bogaerts A (2016) CO2 hydrogenation in a dielectric barrier discharge plasma revealed. J Phys Chem C 120(44):25210–25224. https://doi.org/10.1021/acs.jpcc.6b07639
Kogelschatz U (2003) Dielectric-barrier discharges: their history, discharge physics, and industrial applications. Plasma Chem Plasma Process 23(1):1–46. https://doi.org/10.1023/A:1022470901385
Di L, Zhang J, Zhang X (2018) A review on the recent progress, challenges, and perspectives of atmospheric-pressure cold plasma for preparation of supported metal catalysts. Plasma Process Polym 15(5):1700234. https://doi.org/10.1002/ppap.201700234
Subedi DP, Joshi UM, Wong CS (2017) Dielectric barrier discharge (DBD) plasmas and their applications. In: Rawat RS (ed) Plasma science and technology for emerging economies: an AAAPT experience. Springer, Singapore, pp 693–737. https://doi.org/10.1007/978-981-10-4217-1_13
Niu G, Knodel A, Burhenn S, Brandt S, Franzke J (2021) Review: miniature dielectric barrier discharge (DBD) in analytical atomic spectrometry. Anal Chim Acta 1147:211–239. https://doi.org/10.1016/j.aca.2020.11.034
Bacariza MC, Biset-Peiró M, Graça I, Guilera J, Morante J, Lopes JM, Andreu T, Henriques C (2018) DBD plasma-assisted CO2 methanation using zeolite-based catalysts: structure composition-reactivity approach and effect of Ce as promoter. J CO2 Util 26:202–211. https://doi.org/10.1016/j.jcou.2018.05.013
Lee CJ, Lee DH, Kim T (2017) Enhancement of methanation of carbon dioxide using dielectric barrier discharge on a ruthenium catalyst at atmospheric conditions. Catal Today 293–294:97–104. https://doi.org/10.1016/j.cattod.2017.01.022
Eliasson B, Kogelschatz U, Xue B, Zhou L-M (1998) Hydrogenation of carbon dioxide to methanol with a discharge-activated Catalyst. Ind Eng Chem Res 37(8):3350–3357. https://doi.org/10.1021/ie9709401
Tu X, Verheyde B, Corthals S, Paulussen S, Sels BF (2011) Effect of packing solid material on characteristics of helium dielectric barrier discharge at atmospheric pressure. Phys Plasmas 18(8):080702. https://doi.org/10.1063/1.3619822
Nizio M, Albarazi A, Cavadias S, Amouroux J, Galvez ME, Da Costa P (2016) Hybrid plasma-catalytic methanation of CO2 at low temperature over ceria zirconia supported Ni catalysts. Int J Hydrogen Energy 41(27):11584–11592. https://doi.org/10.1016/j.ijhydene.2016.02.020
Nizio M, Benrabbah R, Krzak M, Debek R, Motak M, Cavadias S, Gálvez ME, Da Costa P (2016) Low temperature hybrid plasma-catalytic methanation over Ni-Ce-Zr hydrotalcite-derived catalysts. Catal Commun 83:14–17. https://doi.org/10.1016/j.catcom.2016.04.023
Zeng Y, Tu X (2017) Plasma-catalytic hydrogenation of CO2 for the cogeneration of CO and CH4 in a dielectric barrier discharge reactor: effect of argon addition. J Phys D Appl Phys 50(18):184004. https://doi.org/10.1088/1361-6463/aa64bb
Mora EY, Sarmiento A, Vera E (2016) Alumina and quartz as dielectrics in a dielectric barrier discharges DBD system for CO2 hydrogenation. J Phys Conf Ser 687(1):012020. https://doi.org/10.1088/1742-6596/687/1/012020
Ahmad F, Lovell EC, Masood H, Cullen PJ, Ostrikov KK, Scott JA, Amal R (2020) Low-temperature CO2 methanation: synergistic effects in plasma-Ni Hybrid catalytic system. ACS Sustain Chem Eng 8(4):1888–1898. https://doi.org/10.1021/acssuschemeng.9b06180
Bogaerts A, Neyts EC, Guaitella O, Murphy AB (2022) Foundations of plasma catalysis for environmental applications. Plasma Sources Sci Technol 31(5):053002. https://doi.org/10.1088/1361-6595/ac5f8e
Ronda-Lloret M, Wang Y, Oulego P, Rothenberg G, Tu X, Shiju NR (2020) CO2 hydrogenation at atmospheric pressure and low temperature using plasma-enhanced catalysis over supported cobalt oxide catalysts. ACS Sustain Chem Eng 8(47):17397–17407. https://doi.org/10.1021/acssuschemeng.0c05565
Di L, Zhang J, Zhang X, Wang H, Li H, Li Y, Bu D (2021) Cold plasma treatment of catalytic materials: a review. J Phys D Appl Phys 54(33):333001. https://doi.org/10.1088/1361-6463/ac0269
Li Q, Zhu X, Yang J, Yu Q, Zhu X, Chu J, Du Y, Wang C, Hua Y, Li H, Xu H (2020) Plasma treated Bi2WO6 ultrathin nanosheets with oxygen vacancies for improved photocatalytic CO2 reduction. Inorg Chem Front 7(3):597–602. https://doi.org/10.1039/C9QI01370A
Zhong K, Zhou A, Zhou G, Li Q, Yang J, Wang Z, Zhu X, Qian J, Hua Y, Li H, Xu H (2021) Plasma-induced black bismuth tungstate as a photon harvester for photocatalytic carbon dioxide conversion. New J Chem 45(4):1993–2000. https://doi.org/10.1039/D0NJ05082B
Horlyck J, Nashira A, Lovell E, Daiyan R, Bedford N, Wei Y, Amal R, Scott J (2019) Plasma treating mixed metal oxides to improve oxidative performance via defect generation. Materials (Basel). https://doi.org/10.3390/ma12172756
Chen X, Zhai X, Hou J, Cao H, Yue X, Li M, Chen L, Liu Z, Ge G, Guo X (2021) Tunable nitrogen-doped delaminated 2D MXene obtained by NH3/Ar plasma treatment as highly efficient hydrogen and oxygen evolution reaction electrocatalyst. Chem Eng J 420:129832–129832. https://doi.org/10.1016/j.cej.2021.129832
Jantarang S, Ligori S, Horlyck J, Lovell EC, Tan TH, Xie B, Amal R, Scott J (2021) Plasma-induced catalyst support defects for the photothermal methanation of carbon dioxide. Materials 14(15):4195. https://doi.org/10.3390/ma14154195
Pastor-Pérez L, Belda-Alcázar V, Marini C, Pastor-Blas MM, Sepúlveda-Escribano A, Ramos-Fernandez EV (2018) Effect of cold Ar plasma treatment on the catalytic performance of Pt/CeO2 in water-gas shift reaction (WGS). Appl Catal B 225:121–127. https://doi.org/10.1016/j.apcatb.2017.11.065
Mikhail M, Da Costa P, Amouroux J, Cavadias S, Tatoulian M, Gálvez ME, Ognier S (2021) Tailoring physicochemical and electrical properties of Ni/CeZrOx doped catalysts for high efficiency of plasma catalytic CO2 methanation. Appl Catal B 294:120233–120233. https://doi.org/10.1016/j.apcatb.2021.120233
Ge Y, He T, Han D, Li G, Zhao R, Wu J (2019) Plasma-assisted CO2 methanation: effects on the low-temperature activity of an Ni–Ce catalyst and reaction performance. R Soc Open Sci. https://doi.org/10.1098/rsos.190750
Pan Q, Peng J, Sun T, Wang S, Wang S (2014) Insight into the reaction route of CO2 methanation: promotion effect of medium basic sites. Catal Commun 45:74–78. https://doi.org/10.1016/j.catcom.2013.10.034
Benrabbah R, Cavaniol C, Liu H, Ognier S, Cavadias S, Gálvez ME, Da Costa P (2017) Plasma DBD activated ceria-zirconia-promoted Ni-catalysts for plasma catalytic CO2 hydrogenation at low temperature. Catal Commun 89:73–76. https://doi.org/10.1016/j.catcom.2016.10.028
Jwa E, Mok YS, Lee SB (2011) Nonthermal plasma-assisted catalytic methanation of CO and CO2 over nickel-loaded alumina. WIT Trans Ecol Environ 143:361–368. https://doi.org/10.2495/ESUS110311
Xu W, Zhang X, Dong M, Zhao J, Di L (2019) Plasma-assisted Ru/Zr-MOF catalyst for hydrogenation of CO2 to methane. Plasma Sci Technol. https://doi.org/10.1088/2058-6272/aaf9d2
Lan L, Wang A, Wang Y (2019) CO2 hydrogenation to lower hydrocarbons over ZSM-5-supported catalysts in a dielectric-barrier discharge plasma reactor. Catal Commun 130:105761–105761. https://doi.org/10.1016/j.catcom.2019.105761
Jwa E, Lee SB, Lee HW, Mok YS (2013) Plasma-assisted catalytic methanation of CO and CO2 over Ni–zeolite catalysts. Fuel Process Technol 108:89–93. https://doi.org/10.1016/j.fuproc.2012.03.008
Wang J, Wang X, AlQahtani MS, Knecht SD, Bilén SG, Chu W, Song C (2023) Synergetic effect of non-thermal plasma and supported cobalt catalyst in plasma-enhanced CO2 hydrogenation. Chem Eng J. https://doi.org/10.1016/j.cej.2022.138661
Xu S, Chansai S, Xu S, Stere CE, Jiao Y, Yang S, Hardacre C, Fan X (2020) CO poisoning of Ru catalysts in CO2 hydrogenation under thermal and plasma conditions: a combined kinetic and diffuse reflectance infrared fourier transform spectroscopy-mass spectrometry study. ACS Catal 10(21):12828–12840. https://doi.org/10.1021/acscatal.0c03620
Chen H, Goodarzi F, Mu Y, Chansai S, Mielby JJ, Mao B, Sooknoi T, Hardacre C, Kegnæs S, Fan X (2020) Effect of metal dispersion and support structure of Ni/silicalite-1 catalysts on non-thermal plasma (NTP) activated CO2 hydrogenation. Appl Catal B 272:119013–119013. https://doi.org/10.1016/j.apcatb.2020.119013
Dębek R, Azzolina-Jury F, Travert A, Maugé F, Thibault-Starzyk F (2019) Low-pressure glow discharge plasma-assisted catalytic CO2 hydrogenation—The effect of metal oxide support on the performance of the Ni-based catalyst. Catal Today 337:182–194. https://doi.org/10.1016/j.cattod.2019.03.039
Chen H, Mu Y, Shao Y, Chansai S, Xiang H, Jiao Y, Hardacre C, Fan X (2020) Nonthermal plasma (NTP) activated metal–organic frameworks (MOFs) catalyst for catalytic CO2 hydrogenation. AIChE J 66(4):1–11. https://doi.org/10.1002/aic.16853
Amouroux J, Cavadias S (2017) Electrocatalytic reduction of carbon dioxide under plasma DBD process. J Phys D Appl Phys 50(46):465501–465501. https://doi.org/10.1088/1361-6463/aa8b56
Biset-Peiró M, Guilera J, Zhang T, Arbiol J, Andreu T (2019) On the role of ceria in Ni-Al2O3 catalyst for CO2 plasma methanation. Appl Catal A 575:223–229. https://doi.org/10.1016/j.apcata.2019.02.028
Hasrack G, Bacariza MC, Henriques C, Da Costa P (2021) On the effect of cobalt promotion over Ni/CeO2 catalyst for CO2 thermal and plasma assisted methanation. Catalysts 12(1):36. https://doi.org/10.3390/catal12010036
Wierzbicki D, Moreno MV, Ognier S, Motak M, Grzybek T, Da Costa P, Gálvez ME (2020) Ni-Fe layered double hydroxide derived catalysts for non-plasma and DBD plasma-assisted CO2 methanation. Int J Hydrogen Energy 45(17):10423–10432. https://doi.org/10.1016/j.ijhydene.2019.06.095
Biset-Peiró M, Mey R, Guilera J, Andreu T (2020) Adiabatic plasma-catalytic reactor configuration: energy efficiency enhancement by plasma and thermal synergies on CO2 methanation. Chem Eng J 393:124786–124786. https://doi.org/10.1016/j.cej.2020.124786
Mikhail M, Wang B, Jalain R, Cavadias S, Tatoulian M, Ognier S, Gálvez ME, Da Costa P (2019) Plasma-catalytic hybrid process for CO2 methanation: optimization of operation parameters. React Kinet Mech Catal 126(2):629–643. https://doi.org/10.1007/s11144-018-1508-8
Gao Y, Dou L, Zhang S, Zong L, Pan J, Hu X, Sun H, Ostrikov K, Shao T (2021) Coupling bimetallic Ni-Fe catalysts and nanosecond pulsed plasma for synergistic low-temperature CO2 methanation. Chem Eng J 420(P2):127693–127693. https://doi.org/10.1016/j.cej.2020.127693
Mikhail M, Da Costa P, Cavadias S, Tatoulian M, Ognier S, Galvez ME (2021) Nickel supported modified zirconia catalysts for CO2 methanation in DBD plasma catalytic hybrid process. Materials Science Forum 1016 MSF:894–899. doi:https://doi.org/10.4028/www.scientific.net/MSF.1016.894
Chen H, Mu Y, Shao Y, Chansai S, Xu S, Stere CE, Xiang H, Zhang R, Jiao Y, Hardacre C, Fan X (2019) Coupling non-thermal plasma with Ni catalysts supported on BETA zeolite for catalytic CO2 methanation. Catal Sci Technol 9(15):4135–4145. https://doi.org/10.1039/c9cy00590k
Guo W, Chen H (2022) Mechanochemical synthesis of Ni–Y/CeO2 catalyst for nonthermal plasma catalytic CO2 methanation. Ind Eng Chem Res 61(4):1666–1674. https://doi.org/10.1021/acs.iecr.1c04456
Ullah N, Su M, Yang Y, Li Z (2023) Enhanced CO2 hydrogenation to light hydrocarbons on Ni-based catalyst by DBD plasma. Int J Hydrogen Energy. https://doi.org/10.1016/j.ijhydene.2023.03.006
Wang J, AlQahtani MS, Wang X, Knecht SD, Bilén SG, Song C, Chu W (2021) One-step plasma-enabled catalytic carbon dioxide hydrogenation to higher hydrocarbons: significance of catalyst-bed configuration. Green Chem 23(4):1642–1647. https://doi.org/10.1039/d0gc03779f
Chen H, Guo W, Fan X (2022) Mechanochemical synthesis of bimetallic NiCo supported on a CeO2 catalyst with less metal loading for non-thermal plasma catalytic CO2 hydrogenation. ACS Eng Au. https://doi.org/10.1021/acsengineeringau.2c00032
Li Y, Zhao J, Bu D, Zhang X, Peng T, Di L, Zhang X (2021) Plasma-assisted Co/Zr-metal organic framework catalysis of CO2 hydrogenation: Influence of Co precursors. Plasma Sci Technol. https://doi.org/10.1088/2058-6272/abeed9
Xu S, Chansai S, Shao Y, Xu S, Yc W, Haigh S, Mu Y, Jiao Y, Stere CE, Chen H, Fan X, Hardacre C (2020) Mechanistic study of non-thermal plasma assisted CO2 hydrogenation over Ru supported on MgAl layered double hydroxide. Appl Catal B 268:118752–118752. https://doi.org/10.1016/j.apcatb.2020.118752
Xu W, Dong M, Di L, Zhang X (2019) A facile method for preparing UiO-66 encapsulated Ru catalyst and its application in plasma-assisted CO2 methanation. Nanomaterials. https://doi.org/10.3390/nano9101432
Li J, Sun Y, Wang B, Xiao H, Wu J, Chen L, Fu M, Ye D (2019) Effect of plasma on catalytic conversion of CO2 with hydrogen over Pd/ZnO in a dielectric barrier discharge reactor. J Phys D Appl Phys 52(24):244001–244001. https://doi.org/10.1088/1361-6463/ab111b
Sun Y, Wu J, Wang Y, Li J, Wang N, Harding J, Mo S, Chen L, Chen P, Fu M, Ye D, Huang J, Tu X (2022) Plasma-catalytic CO2 hydrogenation over a Pd/ZnO catalyst: in situ probing of gas-phase and surface reactions. JACS Au 2(8):1800–1810. https://doi.org/10.1021/jacsau.2c00028
Oshima K, Shinagawa T, Nogami Y, Manabe R, Ogo S, Sekine Y (2014) Low temperature catalytic reverse water gas shift reaction assisted by an electric field. Catal Today 232:27–32. https://doi.org/10.1016/j.cattod.2013.11.035
Wang B, Wang N, Sun Y, Xiao H, Fu M, Li S, Liang H, Qiao Z, Ye D (2023) Dielectric barrier discharge plasma modified Pt/CeO2 catalysts for toluene oxidation: effect of discharge time. Appl Surf Sci 614:156162. https://doi.org/10.1016/j.apsusc.2022.156162
Fan Z, Sun K, Rui N, Zhao B, Liu C-j (2015) Improved activity of Ni/MgAl2O4 for CO2 methanation by the plasma decomposition. J Energy Chem 24(5):655–659. https://doi.org/10.1016/j.jechem.2015.09.004
Mikhail M, Costa PD, Amouroux J, Cavadias S, Tatoulian M, Ognier S, Gálvez ME (2021) Effect of Na and K impurities on the performance of Ni/CeZrOx catalysts in DBD plasma-catalytic CO2 methanation. Fuel 306:121639. https://doi.org/10.1016/j.fuel.2021.121639
Jahanbakhsh MR, Taghvaei H, Khalifeh O, Ghanbari M, Rahimpour MR (2020) Low-temperature CO2 splitting in a noncatalytic dielectric-barrier discharge plasma: effect of operational parameters with a new strategy of experimentation. Energy Fuels 34(11):14321–14332. https://doi.org/10.1021/acs.energyfuels.0c02116
Mok YS, Kang HC, Lee HJ, Koh DJ, Shin DN (2010) Effect of nonthermal plasma on the methanation of carbon monoxide over nickel catalyst. Plasma Chem Plasma Process 30(4):437–447. https://doi.org/10.1007/s11090-010-9231-x
Zou N, Chen J, Qiu T, Zheng Y (2023) Direct hydrogenation of CO2 to ethanol at ambient conditions using Cu(i)-MOF in a dielectric barrier discharge plasma reactor. J Mater Chem A. https://doi.org/10.1039/D3TA00314K
Valdivia-Barrientos R, Pacheco-Sotelo J, Pacheco-Pacheco M, Benítez-Read JS, López-Callejas R (2006) Analysis and electrical modelling of a cylindrical DBD configuration at different operating frequencies. Plasma Sources Sci Technol 15(2):237. https://doi.org/10.1088/0963-0252/15/2/008
Kano M, Satoh G, Iizuka S (2012) Reforming of carbon dioxide to methane and methanol by electric impulse low-pressure discharge with hydrogen. Plasma Chem Plasma Process 32(2):177–185. https://doi.org/10.1007/s11090-011-9333-0
Song HK, Lee H, Choi JW, Na BK (2004) Effect of electrical pulse forms on the CO2 reforming of methane using atmospheric dielectric barrier discharge. Plasma Chem Plasma Process 24(1):57–72. https://doi.org/10.1023/B:PCPP.0000004882.33117.42
Jahanmiri A, Rahimpour MR, Mohamadzadeh Shirazi M, Hooshmand N, Taghvaei H (2012) Naphtha cracking through a pulsed DBD plasma reactor: effect of applied voltage, pulse repetition frequency and electrode material. Chem Eng J 191:416–425. https://doi.org/10.1016/j.cej.2012.02.031
Men YL, Liu Y, Wang Q, Luo ZH, Shao S, Li YB, Pan YX (2019) Highly dispersed Pt-based catalysts for selective CO2 hydrogenation to methanol at atmospheric pressure. Chem Eng Sci 200:167–175. https://doi.org/10.1016/j.ces.2019.02.004
van’t Veer K, Engelmann Y, Reniers F, Bogaerts A (2020) Plasma-catalytic ammonia synthesis in a DBD Plasma: role of microdischarges and their afterglows. J Phys Chem C 124(42):22871–22883. https://doi.org/10.1021/acs.jpcc.0c05110
Yu Q, Kong M, Liu T, Fei J, Zheng X (2012) Characteristics of the decomposition of CO2 in a dielectric packed-bed plasma reactor. Plasma Chem Plasma Process 32(1):153–163. https://doi.org/10.1007/s11090-011-9335-y
Michielsen I, Uytdenhouwen Y, Pype J, Michielsen B, Mertens J, Reniers F, Meynen V, Bogaerts A (2017) CO2 dissociation in a packed bed DBD reactor: first steps towards a better understanding of plasma catalysis. Chem Eng J 326:477–488. https://doi.org/10.1016/j.cej.2017.05.177
Xu S, Chen H, Hardacre C, Fan X (2021) Non-thermal plasma catalysis for CO2 conversion and catalyst design for the process. J Phys D Appl Phys 54(23):233001–233001. https://doi.org/10.1088/1361-6463/abe9e1
Mei D, Zhu X, He Y-L, Yan JD, Tu X (2015) Plasma-assisted conversion of CO2 in a dielectric barrier discharge reactor: understanding the effect of packing materials. Plasma Sources Sci Technol 24(1):15011–15011. https://doi.org/10.1088/0963-0252/24/1/015011
Zhang K, Mukhriza T, Liu X, Greco PP, Chiremba E (2015) A study on CO2 and CH4 conversion to synthesis gas and higher hydrocarbons by the combination of catalysts and dielectric-barrier discharges. Appl Catal A 502:138–149. https://doi.org/10.1016/j.apcata.2015.06.002
Scapinello M, Martini LM, Tosi P (2014) CO2 hydrogenation by CH4 in a dielectric barrier discharge: catalytic effects of nickel and copper. Plasma Process Polym 11(7):624–628. https://doi.org/10.1002/ppap.201400023
Wang L, Yi Y, Guo H, Tu X (2018) Atmospheric pressure and room temperature synthesis of methanol through plasma-catalytic hydrogenation of CO2. ACS Catal 8(1):90–100. https://doi.org/10.1021/acscatal.7b02733
Mei D, Tu X (2017) Conversion of CO2 in a cylindrical dielectric barrier discharge reactor: effects of plasma processing parameters and reactor design. J CO2 Util 19:68–78. https://doi.org/10.1016/j.jcou.2017.02.015
Lu N, Zhang C, Shang K, Jiang N, Li J, Wu Y (2019) Corrigendum: Dielectric barrier discharge plasma assisted CO2 conversion: understanding the effects of reactor design and operating parameters. J Phys D Appl Phys 52:224003. https://doi.org/10.1088/1361-6463/ab0ebb
Wu P, Li X, Ullah N, Li Z (2021) Synergistic effect of catalyst and plasma on CO2 decomposition in a dielectric barrier discharge plasma reactor. Mol Catal 499:111304–111304. https://doi.org/10.1016/j.mcat.2020.111304
Liu J, Zhu X, Hu X, Zhang F, Tu X (2022) Plasma-assisted ammonia synthesis in a packed-bed dielectric barrier discharge reactor: effect of argon addition. Vacuum 197:110786–110786. https://doi.org/10.1016/j.vacuum.2021.110786
Yuan H, Zhu X, Han J, Wang H, Ge Q (2018) Rhenium-promoted selective CO2 methanation on Ni-based catalyst. J CO2 Util 26:8–18. https://doi.org/10.1016/j.jcou.2018.04.010
Lim KH, Yue Y, Bella GX, Zhang T, Hu F, Das S, Kawi S (2023) Sustainable hydrogen and ammonia technologies with nonthermal plasma catalysis: mechanistic insights and technoeconomic analysis. ACS Sustain Chem Eng 11(13):4903–4933. https://doi.org/10.1021/acssuschemeng.2c06515
Parastaev A, Kosinov N, Hensen EJM (2021) Mechanistic study of catalytic CO2 hydrogenation in a plasma by operando DRIFT spectroscopy. J Phys D Appl Phys 54(26):264004. https://doi.org/10.1088/1361-6463/abeb96
Toko S, Ideguchi M, Hasegawa T, Okumura T, Kamataki K, Takenaka K, Koga K, Shiratani M, Setsuhara Y (2022) Effect of gas flow rate and discharge volume on CO2 methanation with plasma catalysis. Jpn J Appl Phys 61:SI1002. https://doi.org/10.35848/1347-4065/ac4822
Xu S, Chansai S, Stere C, Inceesungvorn B, Goguet A, Wangkawong K, Taylor SFR, Al-Janabi N, Hardacre C, Martin PA, Fan X (2019) Sustaining metal–organic frameworks for water–gas shift catalysis by non-thermal plasma. Nat Catal 2(2):142–148. https://doi.org/10.1038/s41929-018-0206-2
Parastaev A, Hoeben WFLM, van Heesch BEJM, Kosinov N, Hensen EJM (2018) Temperature-programmed plasma surface reaction: an approach to determine plasma-catalytic performance. Appl Catal B 239:168–177. https://doi.org/10.1016/j.apcatb.2018.08.011
Azzolina-Jury F, Thibault-Starzyk F (2017) Mechanism of low pressure plasma-assisted CO2 hydrogenation over Ni-USY by microsecond time-resolved FTIR spectroscopy. Top Catal 60(19–20):1709–1721. https://doi.org/10.1007/s11244-017-0849-2
Pitchford LC, Alves LL, Bartschat K, Biagi SF, Bordage M-C, Bray I, Brion CE, Brunger MJ, Campbell L, Chachereau A, Chaudhury B, Christophorou LG, Carbone E, Dyatko NA, Franck CM, Fursa DV, Gangwar RK, Guerra V, Haefliger P, Hagelaar GJM, Hoesl A, Itikawa Y, Kochetov IV, McEachran RP, Morgan WL, Napartovich AP, Puech V, Rabie M, Sharma L, Srivastava R, Stauffer AD, Tennyson J, de Urquijo J, van Dijk J, Viehland LA, Zammit MC, Zatsarinny O, Pancheshnyi S (2017) LXCat: an open-access, web-based platform for data needed for modeling low temperature plasmas. Plasma Processes Polym 14(1–2):1600098. https://doi.org/10.1002/ppap.201600098
Treanor CE, Rich JW, Rehm RG (2003) Vibrational relaxation of anharmonic oscillators with exchange-dominated collisions. J Chem Phys 48(4):1798–1807. https://doi.org/10.1063/1.1668914
Mu Y, Xu S, Shao Y, Chen H, Hardacre C, Fan X (2020) Kinetic study of nonthermal plasma activated catalytic CO2 hydrogenation over Ni supported on silica catalyst. Ind Eng Chem Res 59(20):9478–9487. https://doi.org/10.1021/acs.iecr.0c01477
Kim J, Go DB, Hicks JC (2017) Synergistic effects of plasma–catalyst interactions for CH4 activation. Phys Chem Chem Phys 19(20):13010–13021. https://doi.org/10.1039/C7CP01322A
Neyts EC, Bogaerts A (2014) Understanding plasma catalysis through modelling and simulation—a review. J Phys D Appl Phys 47(22):224010. https://doi.org/10.1088/0022-3727/47/22/224010
Berthelot A, Bogaerts A (2017) Modeling of CO2 plasma: effect of uncertainties in the plasma chemistry. Plasma Sources Sci Technol 26(11):115002–115002. https://doi.org/10.1088/1361-6595/aa8ffb
Alliati M, Mei D, Tu X (2018) Plasma activation of CO2 in a dielectric barrier discharge: a chemical kinetic model from the microdischarge to the reactor scales. J CO2 Util 27:308–319. https://doi.org/10.1016/j.jcou.2018.07.018
Yu J, Zeng Y, Jin Q, Lin W, Lu X (2022) Hydrogenation of CO2 to methane over a Ru/RuTiO2 surface: a DFT investigation into the significant role of the RuO2 overlayer. ACS Catal 12(23):14654–14666. https://doi.org/10.1021/acscatal.2c04539
Sterk EB, Nieuwelink A-E, Monai M, Louwen JN, Vogt ETC, Filot IAW, Weckhuysen BM (2022) Structure sensitivity of CO2 conversion over nickel metal nanoparticles explained by micro-kinetics simulations. JACS Au 2(12):2714–2730. https://doi.org/10.1021/jacsau.2c00430
Dou L, Liu Y, Gao Y, Li J, Hu X, Zhang S, Ostrikov K, Shao T (2022) Disentangling metallic cobalt sites and oxygen vacancy effects in synergistic plasma-catalytic CO2/CH4 conversion into oxygenates. Appl Catal B 318:121830. https://doi.org/10.1016/j.apcatb.2022.121830
Ren J, Guo H, Yang J, Qin Z, Lin J, Li Z (2015) Insights into the mechanisms of CO2 methanation on Ni(111) surfaces by density functional theory. Appl Surf Sci 351:504–516. https://doi.org/10.1016/j.apsusc.2015.05.173
Kim D-Y, Ham H, Chen X, Liu S, Xu H, Lu B, Furukawa S, Kim H-H, Takakusagi S, Sasaki K, Nozaki T (2022) Cooperative catalysis of vibrationally excited CO2 and alloy catalyst breaks the thermodynamic equilibrium limitation. J Am Chem Soc 144(31):14140–14149. https://doi.org/10.1021/jacs.2c03764
Cui Z, Meng S, Yi Y, Jafarzadeh A, Li S, Neyts EC, Hao Y, Li L, Zhang X, Wang X, Bogaerts A (2022) Plasma-catalytic methanol synthesis from CO2 hydrogenation over a supported Cu cluster catalyst: insights into the reaction mechanism. ACS Catal 12(2):1326–1337. https://doi.org/10.1021/acscatal.1c04678
Chen Y, Peng Y, Qian M, Liu S, Zhang J, Wang D (2022) A zero-dimensional model for atmospheric non-thermal plasma CO2hydrogenation: insights into the reaction mechanism. Jpn J Appl Phys. https://doi.org/10.35848/1347-4065/ac79eb
Michiels R, Engelmann Y, Bogaerts A (2020) Plasma catalysis for CO2 hydrogenation: unlocking new pathways toward CH3OH. J Phys Chem C 124(47):25859–25872. https://doi.org/10.1021/acs.jpcc.0c07632
De Bie C, van Dijk J, Bogaerts A (2015) The dominant pathways for the conversion of methane into oxygenates and syngas in an atmospheric pressure dielectric barrier discharge. J Phys Chem C 119(39):22331–22350. https://doi.org/10.1021/acs.jpcc.5b06515
Liao Y, Zhong W, Qian M, Liu S, Zhang J, Wang D (2020) Numerical study on the reaction mechanism of CO2 hydrogenation in atmospheric-pressure dielectric barrier discharge. J Appl Phys. https://doi.org/10.1063/5.0028174
Khunda D, Li S, Cherkasov N, Rishard MZM, Chaffee AL, Rebrov EV (2023) Effect of temperature on the CO2 splitting rate in a DBD microreactor. React Chem Eng 8(9):2223–2233. https://doi.org/10.1039/D3RE00113J
Zhao Q, Mao B, Bai X, Zhao J, Chen C, Zhang X, Wei S, Gao Q (2022) Experimental investigation of the discharge and thermal characteristics of an alternating current dielectric-barrier discharge plasma reactor. Appl Therm Eng 217:119276. https://doi.org/10.1016/j.applthermaleng.2022.119276
Wang B, Mikhail M, Galvez ME, Cavadias S, Tatoulian M, Da Costa P, Ognier S (2020) Coupling experiment and simulation analysis to investigate physical parameters of CO2 methanation in a plasma-catalytic hybrid process. Plasma Process Polym 17(9):1900261. https://doi.org/10.1002/ppap.201900261
Li XS, Li YC, Wang LY, Liu JL, Zhu AM (2022) Real-time measurement of axial temperature in a coaxial dielectric barrier discharge reactor and synergistic effect evaluation for in-plasma catalytic CO2 reduction. Plasma Process Polym 19(5):1–8. https://doi.org/10.1002/ppap.202100229
Zhang Y-R, Neyts EC, Bogaerts A (2016) Influence of the material dielectric constant on plasma generation inside catalyst pores. J Phys Chem C 120(45):25923–25934. https://doi.org/10.1021/acs.jpcc.6b09038
Funding
Open access funding provided by CSIRO Library Services. Not applicable.
Author information
Authors and Affiliations
Contributions
SU developed the review structure, collected data and information, wrote the original draft, and edited the manuscript; YG, LD, YL and TS wrote the sections on reaction mechanisms and DFT modelling, and edited the manuscript; YY and ABM provided supervision and project administration, checked and validated the information presented, and edited the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
ABM is an Editor of Plasma Chemistry and Plasma Processing. He played no role in the review of the manuscript.
Ethical Approval
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ullah, S., Gao, Y., Dou, L. et al. Recent Trends in Plasma-Assisted CO2 Methanation: A Critical Review of Recent Studies. Plasma Chem Plasma Process 43, 1335–1383 (2023). https://doi.org/10.1007/s11090-023-10417-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11090-023-10417-9