
Abstract
The brain, long thought to be isolated from the peripheral immune system, is increasingly recognized to be integrated into a systemic immunological network. These conduits of immune–brain interaction and immunosurveillance processes necessitate the presence of complementary immunoregulatory mechanisms, of which brain regulatory T cells (Treg cells) are likely a key facet. Treg cells represent a dynamic population in the brain, with continual influx, specialization to a brain-residency phenotype and relatively rapid displacement by newly incoming cells. In addition to their functions in suppressing adaptive immunity, an emerging view is that Treg cells in the brain dampen down glial reactivity in response to a range of neurological insults, and directly assist in multiple regenerative and reparative processes during tissue pathology. The utility and malleability of the brain Treg cell population make it an attractive therapeutic target across the full spectrum of neurological conditions, ranging from neuroinflammatory to neurodegenerative and even psychiatric diseases. Therapeutic modalities currently under intense development include Treg cell therapy, IL-2 therapy to boost Treg cell numbers and multiple innovative approaches to couple these therapeutics to brain delivery mechanisms for enhanced potency. Here we review the state of the art of brain Treg cell knowledge together with the potential avenues for future integration into medical practice.
Similar content being viewed by others

Introduction
The brain is increasingly recognized to have connections to the immune system. The immunological compartment of the brain includes both tissue-resident immune cells and leukocytes recruited from the blood, even though these immune populations are less prevalent in the brain than in most other organs. The recruited leukocytes include regulatory T (Treg) cells, which are a specialized subset of T cells that have a critical role in maintaining immune tolerance and promoting an anti-inflammatory environment. Defects in the number of Treg cells, or in their suppressive functions, have been implicated in the pathology of many common immunological diseases1, including neuroinflammatory and neurodegenerative conditions2.
Treg cells in the brain need to be considered in the broader context of tissue-resident Treg cells found across other non-lymphoid tissues3. Although primarily found in the blood and lymphoid tissues, Treg cells migrate into non-lymphoid tissues in the steady state, and accumulate under inflammatory conditions. These tissue-resident Treg cells are thought to recognize self-antigens presented in the tissues, resulting in a limited T cell receptor (TCR) repertoire3. Numerous studies have shown that Treg cells play diverse roles and exhibit distinct phenotypes in non-lymphoid tissues, including visceral adipose tissue4, muscle5, the lungs6, the gut7, the skin8 and the liver9. A common phenotype emerges across the tissue Treg cell populations, distinct from that of lymphoid tissue Treg cells, characterized by elevated expression of molecules such as CD69, IL-10, ST2, AREG, KLRG1 and CTLA4 (refs. 3,10). An analogous population of tissue Treg cells is found in the brains of mice11,12 and humans11, with a cellular phenotype distinct from that of circulating Treg cells (Fig. 1). Nonetheless, the brain Treg cell population is not homogeneous. Although the resting naive-like population of Treg cells that is common in the circulation is near-absent in the brain, a diversity of phenotypes are observed within the activated Treg cell population present in the brain. These phenotypes range from Treg cells that show a generic activated phenotype — which can be seen in Treg cells in lymphoid tissues and is characterized by elevated expression levels of canonical Treg cell markers such as CD25, FOXP3 and CTLA4 — to Treg cells that show a tissue-residency phenotype, which is characterized by the expression of the signature ‘residential’ markers observed across multiple tissues — CD69, KLRG1 and ST2 (ref. 10). It has further been proposed that additional, potentially brain-specific, gene expression is found within the brain-resident Treg cell population11. Such genes include those encoding various neuropeptides (for example, neuropeptide Y and preproenkephalin) and neuronal receptors (for example, 5-hydroxytryptamine receptor 7 (5-HT7 receptor) and arginine vasopressin receptor), suggesting potential interactions between Treg cells and the nervous system12. The expression of the transcription factor BLIMP1 also enables Treg cells in the brain to retain their regulatory capacity during inflammatory insult13.

The main identified functional molecules and associated markers for brain regulatory T cells (Treg cells). These molecules have been classified according to whether they are putative effector molecules, molecules involved in cell migration, residency markers, mediators of homeostasis or neuroimmune interface proteins. AREG, amphiregulin; BDNF, brain-derived neurotrophic factor; CCR2, CC-chemokine receptor 2; TCR, T cell receptor.
Interest in these brain Treg cells is driven, in part, by surprising findings in other tissues, where the tissue-resident Treg cells not only control local inflammation but also have roles in tissue homeostasis and repair3. Growing evidence supports the idea that brain Treg cells likewise have a critical role in neuroprotection and neurological repair, and that these activities are relevant to a range of neuroinflammatory, neurodegenerative and even psychiatric conditions. In this Review, we discuss our current understanding of the Treg cell populations found in the brain, focusing on the cellular kinetics of Treg cells entering and residing in the brain, the anti-inflammatory and reparative functions these cells can have, and the likely pathway towards using these properties of Treg cells in the clinic.
Origin, entry and residency of Treg cells in the brain
Origin
Most brain-resident Treg cells are of thymic origin, based on their expression of helios and neuropilin 1 (ref. 11). TCR sequencing of Treg cells isolated from the brains of mice that have experienced ischaemic stroke reveals a limited diversity of TCR clones, including overlapping TCR sequences between different mice12, suggesting thymic selection of Treg cells against a restricted pool of antigens present in brain. In agreement with this, in the experimental autoimmune encephalomyelitis (EAE) mouse model of multiple sclerosis, Treg cells in the central nervous system (CNS) showed over-representation of specific TCR sequences, suggesting oligoclonal expansion of Treg cells against self-antigens present in the CNS14. Independent data suggest that this expansion is based on the incoming Treg cell pool, rather than being induced de novo from conventional autoreactive T cells15. Despite this, the self-antigens recognized by brain Treg cells remain unknown. It is also possible that the TCR repertoire of brain Treg cells changes over time, with, for example, the proposed early neonatal output consisting of distinct specificities16, although the high turnover would dilute any effect of such early events by adulthood (see ‘Residency’ below).
As cells of thymic origin, brain Treg cells need to traffic to their destination organ via the blood. A prerequisite of this brain entry appears to be a prior antigen-stimulation event, likely in lymphoid organs, to impart the trafficking activation status, with TCR-restricted naive-like Treg cells less able to enter the brain, in both physiological11 and pathological conditions12. The brain Treg cell population is dynamic over the life course of mice, with the first detection of these cells occurring around the time of birth and an increase in the population observed with both age and environmental enrichment11. This suggests that the brain-resident Treg cell population may be influenced by various environmental factors and age-related changes, potentially contributing to its functional diversity in different contexts. To date, relatively few studies have compared Treg cells within the brain barrier compartments with those within the brain parenchyma, but a phenotypic analysis of meningeal and parenchymal Treg cells, in mouse and human tissues, found similar residential marker expression11. Although inconclusive, these data are consistent with the barrier compartment and parenchymal cells sharing an origin, being linked through direct migration flows or simply responding to similar microenvironmental signals.
Barrier compartments and Treg cell entry
Compared with tissue Treg cells resident in other organs, brain-destined Treg cells face a daunting task to transition from the bloodstream into tissue residency. Indeed, until the first half of the twentieth century, the brain was considered by many scientists to be immune privileged and lymphocytes were assumed to be excluded from the healthy brain, with the intact blood–brain barrier preventing entry. The exception to this was in the setting of neuroinflammation, such as during multiple sclerosis, where autoreactive T cells entering barrier compartments were licensed for parenchymal entry by local antigen recognition17. This licensing model prompted a reinterpretation of classical experiments showing long-term survival of foreign tissues grafted into the brain parenchyma. Even these classical immunoprivilege experiments demonstrated that prior exposure to the graft tissue in the periphery allowed for rapid rejection of the graft in the brain18, demonstrating the ability of antigen-experienced lymphocytes to enter and function in the brain. The immune privilege of the brain is thus relative and not absolute. The brain entry of Treg cells, as self-reactive T cells, activated in lymphoid organs and capable of recognizing self-antigen in the barrier compartments, is therefore entirely compatible with the observed gated brain immunoprivilege.
Although not impenetrable, the barriers separating the brain from peripheral organs are highly sophisticated and multi-faceted19. Despite this, there are multiple potential migration routes allowing blood-borne cells to enter brain tissue (Fig. 2a). First, at the blood–brain barrier, the parenchyma of the brain is walled off from the perivascular space and brain interfaces by the glia limitans, an additional layer of basement membrane that is supported by astrocytic end-feet. At the brain surface barrier, this glia limitans borders the meningeal layers (dura mater, arachnoid mater and pia mater), which separate the brain from the skull. Analogous structures are present at the other key interface sites, the choroid plexus and cribriform plate. At the blood–cerebrospinal fluid (CSF) barrier, ependymal cells separate the brain parenchyma and the CSF, whereas epithelial cells form a barrier between the CSF and the choroid plexus vasculature. Finally, at the cribriform plate a thinned border structure lines the olfactory neuron bundles that project through the nasal lymphatics. Each of these barrier surfaces can be used for entry by lymphocytes under some circumstances; however, as different subsets of T cells have differential capacity to cross brain barrier surfaces in vitro20, the dominant route used by Treg cells in vivo remains unknown. Furthermore, pathological contexts will change the expression of molecular determinants of adhesion and extravasation, likely changing the relative importance of each route. Regardless of the route, a small number of Treg cells cross over these barriers and are found within the perivascular spaces and barrier compartments (the meninges, choroid plexus/CSF and cribriform plate), and, to a lesser extent, the brain parenchyma11.

a, Four barrier surfaces act as the interface between the brain and the peripheral immune system. These barrier surfaces act as immunological niches, able to host immune cells, and as potential routes for migration further into the brain. b, The blood–brain barrier provides an interface between the brain and circulation. It is formed by tight junctions between endothelial cells lining cerebral capillaries, pericytes and astrocytic end-feet that form the glia limitans. To enter via this route, regulatory T cells (Treg cells) would need to extravasate and reach the perivascular space (1) and then access the brain parenchyma by crossing the glia limitans (2). Treg cells can activate mechanisms of tissue retention and be recruited to inflamed or tumour sites (3). c, The choroid plexus represents the blood–cerebrospinal fluid (CSF) barrier. The choroid plexus consists of an epithelial layer equipped with tight junctions, surrounding a stromal interface that contains fenestrated blood vessels. The ependymal cells are at the interface between the brain parenchyma and the ventricles that are filled with CSF. To enter via this route, Treg cells would need to translocate the choroid plexus epithelium or circumvent via the basal fold (4) and complete their multistep transmigration from the CSF into the brain parenchyma (5). d, At the surface of the brain, the meninges surround the brain and the spinal cord. The meninges are represented by three layers, the dura mater, the arachnoid mater and the pia mater. The dura mater is characterized by the presence of lymphatic vessels and sinuses. Treg cells in the dural lymphatics are drained into the deep cervical lymph nodes (6). The dural sinuses collect blood from cerebral veins. It has been proposed that sinuses drain CSF from the arachnoid villi originating from the subarachnoid space, where most of the meningeal Treg cells reside, although this role has recently been questioned. Direct channels link the bone marrow of the skull to the surface of the arachnoid barrier, providing a potential, but unproven, route for Treg cell entry (7). e, The cribriform plate barrier provides a direct portal from the nasal cavity to the olfactory bulb within the brain for Treg cells to potentially use. Lymphatic vessels run through the cribriform plate along olfactory cranial nerves penetrating the cribriform plate. During inflammation, T cells originating from the mouse nasal-associated lymphoid tissue migrate along sensory axons towards the olfactory bulb (8). Lymphatic vessels near the cribriform plate can drain Treg cells from the CSF (9). f, Candidate molecular mediators of brain Treg migration across the different barrier compartments, with the numbers (1–9) corresponding to the labels in panels b–e. Treg cells are likely to engage different receptor–ligand pairs depending on the route of entry or exit, and these are likely to change between homeostasis and pathological conditions. CCR2, CC-chemokine receptor 2; CXCR4, CXC-chemokine receptor 4; GR, glucocorticoid receptor; TCR, T cell receptor.
It is likely that entry of Treg cells into the brain is a multistep process. The relative paucity of resting naive-like Treg cells, the behaviour of TCR transgenic Treg cells11 and entry rate modelling of parabiosis data10 suggest that an activation stage is required prior to brain entry, presumably within secondary lymphoid organs. Likewise, the activation of the Nur77 reporter for TCR signal engagement within the CD69+ phenotypically tissue-resident fraction of brain Treg cells suggests that secondary in situ antigen recognition occurs and may drive phenotypic differentiation and prolong the dwell time of Treg cells in the brain. The exact site of this secondary engagement with antigen is unknown; however, it is reasonable to consider that the engagement would need to occur anatomically close to the entry site given the highly limited dwell time of the CD69− fraction of Treg cells10. The barrier compartments are thus critical for brain Treg cell entry, being both the site of initial translocation and the probable site for the secondary stimulation likely to be required for extended residency and further migration into the parenchyma. The entry process is likely to differ depending on the entry route, with highly distinct physical and molecular structures present, but insufficient evidence is currently available.
Molecular candidates for Treg cell entry
The molecular mediators of brain Treg cell migration across the different barrier compartments remain largely unknown in the homeostatic state. In the absence of direct experimental evidence for the routes and mechanisms of Treg cell entry, many of the candidate genes for entry via the potential barrier compartments need to be derived from expression patterns of key molecules, be extrapolated from the migration of other leukocyte subsets or be based on migration under different pathological contexts.
Despite being the primary interface of the brain, remarkably little is known about the extent and mediators of lymphocytes crossing from the vasculature directly across the blood–brain barrier under homeostasis (Fig. 2b). Under pathological conditions such as stroke or injury, the damaged vasculature and interface is a major route for lymphocyte entry. CC-chemokine receptor 2 (CCR2), CCR5, CCR7, CXC-chemokine receptor 4 (CXCR4) and sphingosine-1-phosphate receptor are chemotactic receptors that are likely involved at early stages of this migration21,22,23,24,25, with CCR6 and CCR8 gaining importance at later stages following the upregulation of their chemokine ligands by activated glia12.
The blood–CSF barrier (Fig. 2c) has been proposed as a plausible entry point for T cells into the brain, supported by the presence of T cells in the CSF even in healthy individuals26,27. Across the blood–CSF barrier, Treg cells entering from the blood need to cross the choroid plexus epithelial barrier. It has been proposed that immune cells can exit the stroma at the base of the choroid plexus where it folds out from the ventricular wall, without passing any endothelial or epithelial outer brain barrier28,29. During inflammation, this transmigration may be enhanced by ICAM1-mediated and VCAM1-mediated processes. In rodents, these adhesion molecules are expressed on the apical side of choroid plexus epithelial cells30 and upregulated during EAE31. Notably, the number of leukocytes in the CSF in mice correlates with the levels of leukocyte-trafficking molecules expressed (specifically, the levels of ICAM1, CXCL10 and CCL2) in the choroid plexus epithelium, suggesting that the adhesion of inflammatory T cells to the apical side of the epithelial surface increases the leakiness of the tight junctions, enhancing the flow of leukocytes from the choroid plexus stroma to the CSF. Also involved as a negative regulator of migration, although via an unknown mechanism, is glucocorticoid receptor (GR), with knockdown of this receptor on the choroid plexus enhancing the influx of Treg cells32,33. In EAE, infiltrating pro-inflammatory T helper 17 (TH17) cells use CCR6 to follow a gradient of the chemokine CCL20 produced by the choroid plexus33. This allows the inflammatory cells to rapidly distribute into the CSF. As Treg cells also express CCR6, conservation of this migratory pathway makes it a candidate for Treg cell entry during inflammation. Migration into the brain parenchyma would then require a subsequent translocation, from the CSF across the ependymal layer at the ventricular wall, or the glia limitans at the subarachnoid space, similar to what occurs at the brain surface (discussed below).
At the brain surface barrier (Fig. 2d), where the meninges surround the brain, lymphocytes are mostly found in the dura mater layer, just below the bone of the skull. This dura mater layer is, however, further separated from the brain by the arachnoid barrier, which may serve as a substantive barrier for lymphocyte migration into the deeper brain structures. Indeed, functional impairment of the dural lymphatic vessels has surprisingly little effect on neuroinflammation outcomes, and the dura is less responsive to neuroinflammation than proximal brain surfaces34. The leptomeninges/subarachnoid space, by contrast, is more closely aligned with neuroinflammatory events, and inflammation of these regions has a prominent role in neuropathology, for example in the development of cerebral cortical grey matter pathology during progressive multiple sclerosis35. In terms of mechanistic molecules, meningeal T cells, and in particular meningeal Treg cells, use CCR7 to egress from the tissues22; however, the molecules that mediate Treg cell trafficking from the meninges to the brain parenchyma remain unknown34.
Lymphocyte migration across the cribriform plate (Fig. 2e) is poorly understood. Because of discontinuity of the arachnoid membrane near the cribriform plate36, this represents a potentially unrestricted access route to the CSF. Although largely speculative, axonal breaches may provide a potential route for Treg cell migration through the cribriform plate. Entry of Treg cells from the olfactory mucosa to the olfactory bulbs is therefore, potentially, a migration route into other brain regions. Such a route is likely to be of increased importance during inflammation, when inflammatory mediators such as cytokines can directly and indirectly increase blood–brain barrier permeability37. In addition, during inflammation, meningeal lymphatics near the cribriform plate acquire a phenotype that is permissive for crosstalk with plasmacytoid dendritic cells and T cells. This phenotype includes the upregulation of antigen presentation molecules, expression of PDL1 and expression of podoplanin on the lymphatics36, although whether this pathway is shared between conventional T cells and Treg cells remains to be determined.
Finally, it should be noted that the development of a brain tumour alters the entry dynamics of brain Treg cells, driving large accumulations of this otherwise rare cell type. CCL2, CCL22 and CCL28 are upregulated in the tumour environment, with selective involvement of CCR4, CCR8 and CCR10 chemokine receptors for Treg cell recruitment38,39,40,41. The anatomical route for the initial brain entry in these cases is largely unknown.
These different barrier compartments provide a diverse set of potential routes for Treg cells into the brain, although not all have yet been directly demonstrated and additional routes may yet remain to be discovered. Multiple candidates for molecular mediators of Treg cell passage along these routes exist (Fig. 2f), although it should be emphasized that knowledge here is particularly incomplete, and reliant on mechanisms observed in related cell types. Systematic evaluation of Treg cell migration into these compartments, and the influence of pathology and Treg cell activation status on these events, remains a key gap in knowledge for exploration and potential exploitation.
Residency
Although referred to as tissue-resident, the limited data available for brain Treg cells suggest that their residency is highly transitory in nature. Whereas myeloid cells can remain resident in the mouse brain for the life span of the cell, with certain myeloid lineages remaining resident for the entire life span of the organism, Treg cells are only retained in the mouse brain for a period of weeks. Parabiosis studies in mice show rapid host replacement of brain Treg cells during steady-state conditions, with even the phenotypically resident Treg cell population (that is, those cells expressing CD69) having a dwell time of only ~3–4 weeks on average10,11.
The elevated rate of steady-state replacement is consistent with either tissue egress — such as migration of Treg cells through the draining lymphatics of the brain42 — or with high apoptosis rates, consistent with very low local concentrations of IL-2 in the brain, a rate-limiting survival factor for brain Treg cells43. Even when brain IL-2 levels were synthetically supplemented to match those found in other tissues, parabiosis experiments demonstrated unchanged replacement turnover rates of Treg cells from the brain43, suggesting that migration is independent of the number of Treg cells that could be supported by the relevant survival factors present in the brain. Currently, no parabiotic experiments have been performed dissecting all of the barrier compartments, and thus diversity in residency times is possible across these regions. Whereas retention rates for Treg cells in the brain may change in the settings of neuroinflammation and neuropathology, the data currently available are most consistent with a transient residency of Treg cells in the brain that is driven by continual turnover. This dynamic process may play a role in shaping the functional diversity of the brain-resident Treg cell population.
Brain Treg cell functions
Immunological functions of brain Treg cells
The tightly regulated presence, and highly modified phenotype, of brain Treg cells suggests that these cells have biological functions within brain tissue. Of the known functions of Treg cells outside the brain, the prototypical role is that of suppression of adaptive immune responses. Brain Treg cells express all of the identified Treg cell-associated immunosuppressive molecules; indeed, many of these molecules are expressed at higher levels by brain Treg cells than by circulatory Treg cells. It is therefore axiomatic that brain Treg cells should be capable of controlling the same type of adaptive immune responses controlled in the periphery. These classical anti-inflammatory and immunoregulatory roles of brain Treg cells are now well established in a range of inflammatory settings, in particular in models of EAE, and have been reviewed in depth elsewhere44,45. In the homeostatic state it remains an open question as to whether brain Treg cells control local adaptive immune responses. Although consistent with the known functions, the homeostatic numbers of brain-resident Treg cells and brain-resident conventional T cells are both so low that it is plausible that meaningful interactions are rare enough to render such effects negligible. Analysis of the role of brain-resident Treg cells in controlling local adaptive immunity is made more difficult due to the potent role of peripheral Treg cells in controlling peripheral priming of adaptive immune cells that later migrate to the brain.
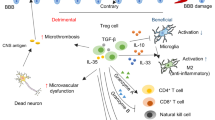
In addition to the suppression of adaptive immunity, brain-resident Treg cells may mediate neuroprotection by suppressing inflammatory gliosis. Both microgliosis and astrogliosis have been implicated in neuroinflammatory damage. Although microglia activation can be reparative, providing for the clearance of apoptotic bodies and debris, excessive activation can result in the production of harmful mediators46. Likewise, the initial response of astrocytes to inflammation has been observed to have beneficial effects, whereas prolonged astrogliosis drives damaging inflammation47 and leaves behind a glial scar that physically interferes with axonal growth48. The ability of brain Treg cells to influence the balance between harmful and reparative glial phenotypes is likely to be a key aspect of neuroprotection49. In a mouse model of Parkinson disease, adoptive transfer of polyclonally activated Treg cells reduced dopaminergic neuronal loss in vivo50. Treg cell-mediated neuroprotection was associated with reduced microglial neurotoxicity and increased levels of brain-derived neurotrophic factor (BDNF) and glial cell line-derived neurotrophic factor (GDNF), which are likely produced by astrocytes, demonstrating that Treg cell administration resulted in phenotypic shifts in both glial populations50. Treg cells have also been shown to inhibit neurotoxic, and promote neuroprotective, microglial phenotypes in an in vivo mouse model of amyotrophic lateral sclerosis51. In a similar manner, Treg cells are protective in mouse models of stroke, with IL-10 production implicated52, AREG shown to be a potential modulator of astrogliosis12 and osteopontin involved in reparative microglial changes53. In a model of traumatic brain injury, osteopontin was also implicated, although in that case the elevated production of osteopontin was indirect, being produced by microglia in Treg cell-rich environments43. Together these data suggest that brain Treg cells have a rich network of anti-inflammatory effects, including direct effects on astrocytes and microglia, that polarize these cells away from destructive inflammatory states, and indirect amplification of these effects through glial-produced effector molecules.
Reparative functions of brain regulatory T cells
Beyond the immunological functions of brain-resident Treg cells, there are emerging data suggesting that these cells have a reparative role. Although difficult to untangle from the anti-inflammatory properties described above, there is growing evidence that brain Treg cells can enhance the survival of neurons following toxic exposures, can drive the regenerative process of remyelination following demyelination and can activate reparative angiogenesis and neurogenesis.
Direct neuroprotective effects of Treg cells have been demonstrated in an in vitro model of neurotoxicity of ventral mesencephalic neurons. Co-culture of primary neurons with activated Treg cells resulted in reduced neuronal cell death following treatment with the neurotoxin 1-methyl-4-phenylpyridinium, an agent used to model the neurotoxicity that occurs in Parkinson disease54. Neuroprotection in this model was contact-mediated and involved interaction between CD47 on Treg cells and signal regulatory protein-α (SIRPα) expressed on ventral mesencephalic neurons, which led to activation of the RAC1–AKT signalling pathway in the neurons54. These results suggest that the neuroprotection observed following Treg cell transfer in a Parkinson disease model may not be purely due to anti-inflammatory properties, but may also be due to Treg cells directly mitigating neurotoxic effects55.
One of the most prominent regenerative roles of Treg cells in the CNS is their support of myelin regeneration. This may be, in part, mediated by their roles in resolving pro-inflammatory, tissue-damaging immune responses and enabling the phagocytic process that is a prerequisite for myelin regeneration56. In addition, however, brain Treg cells directly signal to oligodendrocyte progenitor cells to differentiate into myelin-producing oligodendrocytes, a crucial step in the process of remyelination57. Once differentiated, mature oligodendrocytes will engage with demyelinated axons and re-ensheath these axons with myelin to restore neurological function and neuroprotection58. This capacity of Treg cells to drive oligodendrocyte differentiation and remyelination has been demonstrated directly in animal models of toxin-induced57, autoimmune59 and viral-induced demyelination60, as well as in stroke53. CCN3 production is important57 but not essential61 for this process, with additional molecular mediators potentially including ITGA2 and MCAM influencing oligodendrocyte precursors62, and osteopontin working via microglial reprogramming53. As IGF1 and oncostatin M are also expressed by brain Treg cells53 and aid oligodendrocyte precursor differentiation63,64, they are potential candidates in the process too. This resulting re-ensheathment of neuronal axons may, in turn, aid protection from inflammation, as remyelinated axons are less susceptible to neurodegeneration than demyelinated axons65,66.
Finally, a role has been reported for brain Treg cells in reparative neurogenesis and angiogenesis. In a mouse model of stroke, Treg cells promote neuronal stem cell proliferation, aiding neurogenesis67. This process is reported to be mediated by IL-10 (ref. 67) and galectin 1 (ref. 68), with BDNF another key candidate69. Although evidence at this stage is scarce, Treg cells are associated with preservation of the blood–brain barrier and repair of damaged vasculature. This may be through inhibition of neutrophil70 and microglial cell71 activation, yet effector molecules secreted by brain Treg cells, including IL-10, AREG and galectin 1, are also known to support vascular remodelling72,73, suggesting that a more direct effect may be occurring. Although requiring further exploration, both enhanced neurogenesis and vascular repair are likely non-immunological benefits provided by brain Treg cells.
These results demonstrate the neuroprotective capacity of brain Treg cells through multiple modalities: preventing adaptive immune activation, dampening innate inflammatory responses among resident glia, direct neuroprotection and enhancement of regenerative processes. In many cases, however, the relative contribution of these neuroprotective pathways remains completely unknown. For example, the protective capacities of brain Treg cells in ameliorating cognitive decline in ageing mice74 could be driven through their blockade of harmful CD8+ T cell responses, reprogramming of activated microglia and astrocytes, direct protection of neurons from harmful inflammatory mediators or an improved regenerative pathway75. As primary correction of one aspect is likely to also drive secondary improvements in other read-outs, untangling the causative mode of neuroprotection for brain Treg cells will remain problematic.
Potential limitations to the reparative functions of brain regulatory T cells
Of note, the majority of studies of Treg cells in vascular and neural regeneration focus on Treg cell function in acute phases of the response and at relatively short timeframes after insult. These phases generally feature increased blood–brain barrier permeability and, often, glial scars have not yet formed, facilitating recruitment of Treg cells to damaged tissue. Less attention has been paid to later stages of disease pathogenesis when blood–brain barrier function is restored and glial scars may have developed, potentially impeding the recruitment, access and bioactivity of Treg cells into damaged tissue. Whether Treg cells can still drive tissue regeneration in these settings is not well understood.
Harnessing brain regulatory T cells for advanced therapeutics
Amplification of brain regulatory T cells to reverse neuroinflammation
Exploiting the anti-inflammatory, pro-repair modalities of brain Treg cells has been proposed in a plethora of conditions — including injuries (traumatic brain injury), vascular conditions (stroke), autoimmune diseases (multiple sclerosis), neurodegenerative conditions (Alzheimer disease, Parkinson disease and amyotrophic lateral sclerosis), psychiatric disorders (depression) and the cognitive decline observed in ageing (Fig. 3a). There are two basic modalities of Treg cell therapy that are currently the focus of most attention — cell therapy (Fig. 3b) and IL-2 therapy (Fig. 3c).

Potential beneficial effects of regulatory T (Treg) cell-centric therapies could span neuroinflammatory injuries such as traumatic brain injury and stroke, autoimmune diseases such as multiple sclerosis, neurodegenerative conditions ranging from Alzheimer disease to Parkinson disease and amyotrophic lateral sclerosis, psychiatric disorders such as depression and the cognitive decline experienced in normal ageing. a, There are currently three main Treg cell cellular products developed for cell therapy: polyclonal Treg cells isolated from blood and expanded in vitro using anti-CD3/CD28 antibodies and IL-2 (left); astrocytes used to stimulate Treg cells from the recipient in vitro in the presence of IL-33 and serotonin to confer part of the properties of brain-derived Treg cells (expression of ST2 and 5-HT7) (middle); and expansion of polyclonal engineered Treg cells expressing chimeric antigen receptor (CAR) or artificial T cell receptor (TCR) that recognizes a target antigen (right). b, Mechanism of Treg cell therapy. Treg cells infused via the blood enter the brain. The cell therapy is proposed to reduce pathology by exerting immunosuppressive effects on effector T cells, repolarizing microglia and astrocytes, and enhanced repair processes. Key mediators are likely to vary based on pathology, but may include AREG, IL-10, osteopontin and TGFβ. c, Main modalities of IL-2 therapy. IL-2 as a biologic can be delivered systemically in low doses or can be intrathecally injected in a slow release carrier. Treg cells are highly sensitive to IL-2, accumulating in number (top). Gene delivery systems, such as adeno-associated viral vector (AAV), are capable of crossing an intact blood–brain barrier and delivering the DNA needed for local production in IL-2. Accumulated brain Treg cells rewire the brain into an anti-inflammatory environment (bottom).
Cell therapy for neuroinflammation
Cellular therapies with Treg cells have been carried out in both animal models and clinical trials across peripheral organ transplantation and autoimmunity. The basic premise of Treg cell therapy is that the use of autologous purification, in vitro expansion and, finally, infusion back into the circulation can increase the total number of Treg cells present in the system, reducing inflammation76. Although such approaches can temporarily boost Treg cell numbers, the tight homeostasis control over the total number of Treg cells in the system77 suggests that a polyclonal Treg cell therapy will be best suited for neuroinflammatory conditions where a short-term immunological suppression can lead to long-term clinical benefit. Potential routes to circumvent this limitation include the use of antigen-specific Treg cell expansion, where polyclonal displacement will enrich the repertoire for desired Treg cell TCR specificities, and genomic engineering of Treg cells, imparting biological properties for improved retention, expansion or function (Fig. 3b). In each case, the desired outcome will be an enrichment of brain Treg cells, driving pro-repair and anti-inflammatory processes (Fig. 3c). The latter two approaches have the theoretical benefit of fewer off-target systemic effects, as the approaches are intended to impart brain-specific homing or accumulation.
Two examples serve to demonstrate the potential approaches of advanced Treg cell therapies. In Parkinson disease, preclinical testing has built upon standard Treg cell transfer50 to create an ex vivo Treg cell induction approach that enriches the repertoire for antigen-specific brain Treg cells78. By stimulating naive Treg cells from the spleen in the presence of primary astrocytes, IL-33 and serotonin, the resulting expanded population had transcriptional profiles closer to those observed in bona fide brain Treg cells. This enriched population demonstrated preferential brain entry and more effective control of neurodegeneration in models of Parkinson disease78. In principle, a single enrichment protocol such as this could have broad utility across any neuroinflammatory condition. Whether this protocol can be replicated through co-culture with other brain cells, and the degree to which it mimics the physiological process of in situ differentiation, remains unknown. In multiple sclerosis, one small clinical trial was conducted to examine the potential of Treg cell-based therapy. This phase Ib/IIa clinical trial involved patients with relapsing–remitting multiple sclerosis. Among these participants, 11 individuals received Treg cells that were expanded ex vivo and administered intravenously, whereas 3 individuals received freshly isolated Treg cells delivered intrathecally. None of the participants experienced adverse effects following the treatment. Intriguingly, whereas participants who received Treg cells intravenously experienced disease relapses and increased disability, this was not observed in the three participants who received Treg cells intrathecally79. Although the trial had a limited number of participants, these findings suggest that in individuals with multiple sclerosis it may be the local effects of brain-resident Treg cells on neuroinflammation that are most potent, and therapeutic approaches may need to focus on boosting this tissue-resident population rather than the systemic circulating population.
Relevant to multiple sclerosis, additional studies in the preclinical EAE model have taken an alternative route of genome engineering to drive brain localization of Treg cells. Treg cell transfer experiments had long been known to suppress EAE severity80, although determining whether this was due to brain residency impeding local inflammation or lymph node residency impairing peripheral priming of autoreactive cells has proven difficult. Antigen-specific Treg cells, such as TCR transgenic Treg cells, are more efficient suppressors of EAE. Whereas therapeutic use of TCR transgenesis is still in early clinical development, the use of chimeric antigen receptors (CARs) allows T cells to be imparted with a given antigen specificity, using an antibody fused to the TCR signalling apparatus81. CARs can therefore be used to impart monoclonal-like properties on a polyclonal repertoire, and can be used to create CAR Treg cells either by starting with a polyclonal Treg cell population or by coupling CAR expression with forced expression of FOXP3. Franssen et al. took the latter route, creating myelin oligodendrocyte glycoprotein (MOG)-specific CAR Treg cells82. These cells were highly efficient at entering the brain after intranasal delivery, and impeded the neuroinflammatory EAE response82. A similar approach can transform Treg cells isolated from patients with multiple sclerosis, imparting beneficial properties in EAE83. Although CAR Treg cells face substantial challenges in terms of scalability and cost, the genomic engineering step potentially enables the incorporation of additional design advantages. For example, CAR Treg cells could be equipped with adhesion molecules to improve brain entry, supplemented via the IL-2 pathway to improve longevity, engineered to enhance their production of effector molecules or supplemented through the addition of biosensor pathways to drive activation at inflammatory sites84. Due to these advantages, although the applications may be niche, CAR Treg cells, or the related recombinant TCR Treg cells, are likely to become part of the brain Treg cell therapeutic toolbox.
IL-2 therapy for neuroinflammation
In the biologic space, the most developed therapies for enhancing Treg cell-mediated effects are the IL-2 therapies. IL-2 is a key cytokine for Treg cell homeostasis, setting the cellular niche size through control of proliferation and apoptosis85. The provision of exogenous low-dose IL-2 is safe and well-characterized to drive an expansion of Treg cell numbers, and there is emerging consensus on the utility of multiple low-dose IL-2 treatment for autoimmune diseases86. A potential limitation of this approach is that the delivery of IL-2 systemically drives an expansion of Treg cell populations in the peripheral blood and secondary lymphoid organs, which, although likely to be efficacious in the setting of systemic autoimmunity, may have limited benefits in neuroinflammation (in line with the data on intrathecal versus peripheral Treg cell therapy trials). Nonetheless, a randomized clinical trial (NCT02424396) of low-dose IL-2 in multiple sclerosis has reported promising results, with peripheral Treg cell activation and expansion87. Despite the trial not being powered for efficacy, a trend towards improved clinical outcomes was also observed, where new active brain lesions showed a non-significant tendency to be reduced in patients treated with low-dose IL-2 (ref. 87). These results have inspired a broad investigation of the effects of IL-2 in diseases with an immune component, even beyond the field of autoimmunity. In mouse models, low-dose IL-2 or IL-2–anti-IL-2 antibody complexes show promising results in Alzheimer disease88, amyotrophic lateral sclerosis88,89, migraine90 and depression91, among other neurological conditions. This treatment has proceeded to clinical trials for Alzheimer disease (NCT05821153) and amyotrophic lateral sclerosis (NCT04055623, NCT02059759, NCT03039673, NCT03241784). The amyotrophic lateral sclerosis results have been promising to date. In a trial combining IL-2 with Treg cell infusion, six out of eight patients showed slowed disease progression92. Another small study in patients with amyotrophic lateral sclerosis explored a combination of Treg cells and IL-2. Treg cells expanded ex vivo were administered intravenously concomitantly with low-dose IL-2 injection subcutaneously. The expanded autologous Treg cell therapy suppressed oxidative stress responses and stabilized the clinical status93. Finally, the MIROCALS trial, using low-dose IL-2 in a large cohort of patients with amyotrophic lateral sclerosis, demonstrated safety and a small decrease in death among the treated group with less severe diseases94. The development of engineered forms of IL-2, with improved Treg cell-promoting properties, may further enhance the prospects of direct IL-2 therapy in the neuro-space.
A further potential for improving IL-2 therapy in neurological applications comes from targeted delivery (Fig. 3c). The systemic administration route is well established for the expansion of circulating Treg cells. This expansion likely feeds into the brain Treg cell population via continual recruitment, but diminishing returns will be observed in the brain. Direct delivery of IL-2 into the brain, either through intrathecal or intracerebroventricular injection, allows for brain residency effects to be preferentially gained. Although the half-life of IL-2 is too short for such a delivery of the unmodified biologic to be viable, engineered forms of IL-2 with elevated longevity may be effective95. Alternatively, numerous approaches for slow release or delayed production are being developed, including slow release microspheres96 or occlusion bodies97, lipid-encapsulated IL2 mRNA98 and next-generation nanotechnology approaches99. One promising form of IL-2 delivery is that of gene delivery. By using viral vectors, such as adeno-associated viral vectors (AAVs), the advantages of systemic delivery can be combined with brain-based production (Fig. 3c). The first such approach was an AAV-encoded IL2 vector, using a blood–brain barrier-crossing AAV capsid. Capable of driving both systemic and brain-based IL-2 production, this vector was beneficial in a mouse model of Alzheimer disease100. A refinement of this approach encoded astrocyte-specific production, to eliminate off-target systemic effects, with the capacity to include a small molecule-inducible system43. This approach was beneficial in mouse models of traumatic brain injury, stroke and EAE43. Astrocyte-directed gene delivery was also beneficial in reducing the cognitive decline observed in ageing mice, although not in a mouse model of Alzheimer disease74, suggesting divergent utility of global and brain-restricted IL2 gene delivery in neuroinflammation. Although the focus here has been on innovations in IL-2 therapy, other cytokines such as IL-33 can regulate the number of Treg cells in the brain, albeit to a lesser extent, and may show similar utility10. Furthermore, as more research is performed on brain Treg cells and their effector molecules are characterized (Fig. 1), many of these therapeutic modalities can be adapted to directly deliver the brain Treg cell-associated effector molecules, bypassing the need for IL-2 and Treg cells themselves. Such approaches have further advantages in that they may overcome potential mechanical barriers (such as glial scars) that could limit the entry and/or bioactivity of Treg cells.
Depletion of brain regulatory T cells in cancer
Although the immunosuppressive role of brain-resident Treg cells may provide a boon for the treatment of neuroinflammation, the reverse approach is likely to be needed in the cancer context. In the most common forms of primary cancers of the brain — glioblastomas — Treg cells accumulate to much higher levels than observed in the healthy brain101, with the tumours producing soluble factors capable of enhancing proliferation and recruitment of Treg cells102. The degree to which these resident Treg cells interfere with the spontaneous immunological recognition and clearance of the tumours is debatable, with conflicting evidence on whether Treg cell infiltration predicts survival outcomes101,103. Brain Treg cells, however, do appear to play a key role in the resistance of glioblastoma to immune checkpoint blockade treatment. In mouse models of glioblastoma, tumours are poorly responsive to immune checkpoint blockade involving anti-PD1 antibody treatment, with curtailed expansion of effector CD8+ T cells104,105. Although previously explained by ‘immunological coldness’, these poor outcomes are also explained, in part, by the expansion of brain Treg cells within the glioblastoma. Targeting of these Treg cells through systemic treatment with anti-CD25 antibody to deplete circulating Treg cells and brain-resident Treg cells104, or anti-GITR antibody treatment to convert Treg cells into effector TH1 cells105, allows for improved cytotoxic responses and tumour eradication. In the patient context, it is likely that brain Treg cell depletion will be required in order to improve the disappointing clinical results seen when treating glioblastoma with immune checkpoint blockade. Although numerous different modalities for Treg cell depletion are being developed, one of the more promising avenues is the use of anti-CD25 antibody. An anti-CD25 antibody106 has been trialled in patients with glioblastoma, with tolerable toxicity results and efficient Treg cell depletion observed (NCT04158583). Follow-up clinical trials combining immune checkpoint blockade and Treg cell depletion will determine whether the approach will be successful. This potential breakthrough in treating previously ‘cold’ brain tumours could be further developed through targeted approaches that deplete brain-resident Treg cells while leaving the circulating population intact. Although several promising approaches are being developed to target biologics to a particular tissue, in this particular case the blood–brain barrier may work in favour of treatment: local infusion of Treg cell-depleting agents may end up restricted to the brain, reducing off-target systemic effects. Considering the high mortality and poor therapeutic options for these patients, targeting Treg cells in brain tumours may be a rapidly evolving field with high potential for therapeutic benefit.
Conclusions
Brain Treg cells defy many of our preconceptions of brain cells — they are highly plastic, migratory and transient. The population of brain Treg cells that is resident, temporarily, within the brain is continually replaced by incoming cells, only adopting their brain-based phenotype for a brief sojourn in the tissue. At the same time, the functions of brain Treg cells defy the norms expected of immune cells. Although they possess the same capacity for classical immune suppression as peripheral Treg cells, in the unique immunological environment of the brain the most important role of brain Treg cells may be the unorthodox functions being unearthed: namely, calming down activated glial populations, enhancing the resiliency of neurons and enabling the remyelination function of mature oligodendrocytes. Yet it is these very properties of brain Treg cells, defying the norms of both brain and peripheral immunity, that make brain Treg cells enticing as a therapeutic target. As a transient and dynamic population, continually reseeding the brain from the periphery, brain Treg cells are malleable targets, with the potential to intercede via the circulating population. Likewise, the multi-modality of brain Treg cells, influencing immunity, gliosis, resilience and repair, provides far greater potential therapeutic gain than a single target treatment could achieve. With the potential to harness brain Treg cells directly, via cell therapy, or indirectly, via IL-2 therapy, the active preclinical and clinical trial research in this space is likely to deliver novel therapies for a range of neurological pathologies. Finally, brain Treg cells provide a learning template, as a black box waiting to be opened to identify the molecular mediators that could bypass the need for brain Treg cells entirely and provide their therapeutic potential via as yet to be identified drugs.
References
DuPage, M. & Bluestone, J. A. Harnessing the plasticity of CD4+ T cells to treat immune-mediated disease. Nat. Rev. Immunol. 16, 149–163 (2016).
Machhi, J. et al. Harnessing regulatory T cell neuroprotective activities for treatment of neurodegenerative disorders. Mol. Neurodegener. 15, 32 (2020).
Panduro, M., Benoist, C. & Mathis, D. Tissue Tregs. Annu. Rev. Immunol. 34, 609–633 (2016).
Feuerer, M. et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat. Med. 15, 930–939 (2009).
Burzyn, D. et al. A special population of regulatory T cells potentiates muscle repair. Cell 155, 1282–1295 (2013).
Arpaia, N. et al. A distinct function of regulatory T cells in tissue protection. Cell 162, 1078–1089 (2015).
Schiering, C. et al. The alarmin IL-33 promotes regulatory T-cell function in the intestine. Nature 513, 564–568 (2014).
Rosenblum, M. D. et al. Response to self antigen imprints regulatory memory in tissues. Nature 480, 538–542 (2011).
Delacher, M. et al. Genome-wide DNA-methylation landscape defines specialization of regulatory T cells in tissues. Nat. Immunol. 18, 1160–1172 (2017).
Burton, O. et al. The tissue-resident regulatory T cell pool is shaped by transient multi-tissue migration and a conserved residency program. Preprint at bioRxiv https://doi.org/10.1101/2023.08.14.553196 (2023).
Pasciuto, E. et al. Microglia require CD4 T cells to complete the fetal-to-adult transition. Cell 182, 625–640.e24 (2020). This study is the first to analyse the phenotype, kinetics and function of brain-resident Treg cells in the homeostatic state in mice and humans.
Ito, M. et al. Brain regulatory T cells suppress astrogliosis and potentiate neurological recovery. Nature 565, 246–250 (2019). This study unveils innovative mechanisms through which Treg cells actively participate in the process of tissue repair following brain injury.
Garg, G. et al. Blimp1 prevents methylation of Foxp3 and loss of regulatory T cell identity at sites of inflammation. Cell Rep. 26, 1854–1868.e5 (2019).
O’Connor, R. A., Malpass, K. H. & Anderton, S. M. The inflamed central nervous system drives the activation and rapid proliferation of Foxp3+ regulatory T cells. J. Immunol. 179, 958–966 (2007).
Korn, T. et al. Myelin-specific regulatory T cells accumulate in the CNS but fail to control autoimmune inflammation. Nat. Med. 13, 423–431 (2007).
Yang, S., Fujikado, N., Kolodin, D., Benoist, C. & Mathis, D. Immune tolerance. Regulatory T cells generated early in life play a distinct role in maintaining self-tolerance. Science 348, 589–594 (2015).
Schlager, C. et al. Effector T-cell trafficking between the leptomeninges and the cerebrospinal fluid. Nature 530, 349–353 (2016).
Medawar, P. B. Immunity to homologous grafted skin; the fate of skin homografts transplanted to the brain, to subcutaneous tissue, and to the anterior chamber of the eye. Br. J. Exp. Pathol. 29, 58–69 (1948).
Proulx, S. T. & Engelhardt, B. Central nervous system zoning: how brain barriers establish subdivisions for CNS immune privilege and immune surveillance. J. Intern. Med. 292, 47–67 (2022).
Nishihara, H. et al. Human CD4+ T cell subsets differ in their abilities to cross endothelial and epithelial brain barriers in vitro. Fluids Barriers CNS 17, 3 (2020).
Li, P. et al. C–C chemokine receptor type 5 (CCR5)-mediated docking of transferred Tregs protects against early blood–brain barrier disruption after stroke. J. Am. Heart Assoc. 6, e006387 (2017).
Da Mesquita, S. et al. Aging-associated deficit in CCR7 is linked to worsened glymphatic function, cognition, neuroinflammation, and β-amyloid pathology. Sci. Adv. 7, eabe4601 (2021).
Ben-Yehuda, H. et al. Key role of the CCR2–CCL2 axis in disease modification in a mouse model of tauopathy. Mol. Neurodegener. 16, 39 (2021).
Lucaciu, A. et al. A sphingosine 1-phosphate gradient is linked to the cerebral recruitment of T helper and regulatory T helper cells during acute ischemic stroke. Int. J. Mol. Sci. 21, 6242 (2020).
Lee, H. T. et al. A crucial role of CXCL14 for promoting regulatory T cells activation in stroke. Theranostics 7, 855–875 (2017).
Hrastelj, J. et al. CSF-resident CD4+ T-cells display a distinct gene expression profile with relevance to immune surveillance and multiple sclerosis. Brain Commun. 3, fcab155 (2021).
Kivisakk, P. et al. Human cerebrospinal fluid central memory CD4+ T cells: evidence for trafficking through choroid plexus and meninges via P-selectin. Proc. Natl Acad. Sci. USA 100, 8389–8394 (2003).
Llovera, G. et al. The choroid plexus is a key cerebral invasion route for T cells after stroke. Acta Neuropathol. 134, 851–868 (2017).
Wolburg, H. & Mack, A. F. Comment on the topology of mammalian blood–cerebrospinal fluid barrier. Neurol. Psychiatry Brain Res. 20, 70–72 (2014).
Steffen, B. J., Breier, G., Butcher, E. C., Schulz, M. & Engelhardt, B. ICAM-1, VCAM-1, and MAdCAM-1 are expressed on choroid plexus epithelium but not endothelium and mediate binding of lymphocytes in vitro. Am. J. Pathol. 148, 1819–1838 (1996).
Kunis, G. et al. IFN-γ-dependent activation of the brain’s choroid plexus for CNS immune surveillance and repair. Brain 136, 3427–3440 (2013).
Kertser, A. et al. Corticosteroid signaling at the brain–immune interface impedes coping with severe psychological stress. Sci. Adv. 5, eaav4111 (2019).
Reboldi, A. et al. C–C chemokine receptor 6-regulated entry of TH-17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nat. Immunol. 10, 514–523 (2009).
Li, Z. et al. Blockade of VEGFR3 signaling leads to functional impairment of dural lymphatic vessels without affecting autoimmune neuroinflammation. Sci. Immunol. 8, eabq0375 (2023).
Howell, O. W. et al. Meningeal inflammation is widespread and linked to cortical pathology in multiple sclerosis. Brain 134, 2755–2771 (2011).
Hsu, M. et al. Neuroinflammation creates an immune regulatory niche at the meningeal lymphatic vasculature near the cribriform plate. Nat. Immunol. 23, 581–593 (2022).
Dileepan, T. et al. Group A streptococcus intranasal infection promotes CNS infiltration by streptococcal-specific TH17 cells. J. Clin. Invest. 126, 303–317 (2016).
Jacobs, J. F. et al. Prognostic significance and mechanism of Treg infiltration in human brain tumors. J. Neuroimmunol. 225, 195–199 (2010).
Chang, A. L. et al. CCL2 produced by the glioma microenvironment is essential for the recruitment of regulatory T cells and myeloid-derived suppressor cells. Cancer Res. 76, 5671–5682 (2016).
Jordan, J. T. et al. Preferential migration of regulatory T cells mediated by glioma-secreted chemokines can be blocked with chemotherapy. Cancer Immunol. Immunother. 57, 123–131 (2008).
Facciabene, A. et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and Treg cells. Nature 475, 226–230 (2011).
Louveau, A. et al. Structural and functional features of central nervous system lymphatic vessels. Nature 523, 337–341 (2015). This work presents compelling evidence that lymphatic vessels within the dura mater play a crucial role in transporting antigens originating from the CNS to the cervical lymph node, reshaping our understanding of immune responses within the CNS.
Yshii, L. et al. Astrocyte-targeted gene delivery of interleukin 2 specifically increases brain-resident regulatory T cell numbers and protects against pathological neuroinflammation. Nat. Immunol. 23, 878–891 (2022). This study includes the first immune therapy to expand specifically brain-resident Treg cells without impacting the peripheral compartment.
O’Connor, R. A. & Anderton, S. M. Foxp3+ regulatory T cells in the control of experimental CNS autoimmune disease. J. Neuroimmunol. 193, 1–11 (2008).
Korn, T. Foxp3+ regulatory T cells in the central nervous system and other nonlymphoid tissues. Eur. J. Immunol. 53, e2250227 (2023).
Lyu, J. et al. Microglial/macrophage polarization and function in brain injury and repair after stroke. CNS Neurosci. Ther. 27, 515–527 (2021).
Barres, B. A. The mystery and magic of glia: a perspective on their roles in health and disease. Neuron 60, 430–440 (2008).
Wanner, I. B. et al. A new in vitro model of the glial scar inhibits axon growth. Glia 56, 1691–1709 (2008).
He, X. et al. Programmed death protein 1 is essential for maintaining the anti-inflammatory function of infiltrating regulatory T cells in a murine spinal cord injury model. J. Neuroimmunol. 354, 577546 (2021).
Reynolds, A. D., Banerjee, R., Liu, J., Gendelman, H. E. & Mosley, R. L. Neuroprotective activities of CD4+CD25+ regulatory T cells in an animal model of Parkinson’s disease. J. Leukoc. Biol. 82, 1083–1094 (2007).
Beers, D. R. et al. Endogenous regulatory T lymphocytes ameliorate amyotrophic lateral sclerosis in mice and correlate with disease progression in patients with amyotrophic lateral sclerosis. Brain 134, 1293–1314 (2011).
Liesz, A. et al. Regulatory T cells are key cerebroprotective immunomodulators in acute experimental stroke. Nat. Med. 15, 192–199 (2009).
Shi, L. et al. Treg cell-derived osteopontin promotes microglia-mediated white matter repair after ischemic stroke. Immunity 54, 1527–1542.e8 (2021). This study shows that Treg cell depletion impairs oligodendrogenesis, tissue repair and functional recovery in a mouse model of stroke, with regenerative Treg cell function attributed to promoting reparative microglia via osteopontin.
Huang, Y., Liu, Z., Cao, B. B., Qiu, Y. H. & Peng, Y. P. Treg cells protect dopaminergic neurons against MPP+ neurotoxicity via CD47–SIRPA interaction. Cell Physiol. Biochem. 41, 1240–1254 (2017).
Badr, M. et al. Expansion of regulatory T cells by CD28 superagonistic antibodies attenuates neurodegeneration in A53T-α-synuclein Parkinson’s disease mice. J. Neuroinflammation 19, 319 (2022).
Kotter, M. R., Li, W. W., Zhao, C. & Franklin, R. J. Myelin impairs CNS remyelination by inhibiting oligodendrocyte precursor cell differentiation. J. Neurosci. 26, 328–332 (2006).
Dombrowski, Y. et al. Regulatory T cells promote myelin regeneration in the central nervous system. Nat. Neurosci. 20, 674–680 (2017).
Franklin, R. J. M. & Ffrench-Constant, C. Regenerating CNS myelin — from mechanisms to experimental medicines. Nat. Rev. Neurosci. 18, 753–769 (2017).
McIntyre, L. L. et al. Regulatory T cells promote remyelination in the murine experimental autoimmune encephalomyelitis model of multiple sclerosis following human neural stem cell transplant. Neurobiol. Dis. 140, 104868 (2020).
Plaisted, W. C. et al. Remyelination is correlated with regulatory T cell induction following human embryoid body-derived neural precursor cell transplantation in a viral model of multiple sclerosis. PLoS ONE 11, e0157620 (2016).
de la Vega Gallardo, N. et al. Dynamic CCN3 expression in the murine CNS does not confer essential roles in myelination or remyelination. Proc. Natl Acad. Sci. USA 117, 18018–18028 (2020).
Fuente, A. G. D. L. et al. Ageing impairs the regenerative capacity of regulatory T cells in central nervous system remyelination. Preprint at bioRxiv https://doi.org/10.1101/2023.01.25.525562 (2023).
Hsieh, J. et al. IGF-I instructs multipotent adult neural progenitor cells to become oligodendrocytes. J. Cell Biol. 164, 111–122 (2004).
Glezer, I. & Rivest, S. Oncostatin M is a novel glucocorticoid-dependent neuroinflammatory factor that enhances oligodendrocyte precursor cell activity in demyelinated sites. Brain Behav. Immun. 24, 695–704 (2010).
Irvine, K. A. & Blakemore, W. F. Remyelination protects axons from demyelination-associated axon degeneration. Brain 131, 1464–1477 (2008).
Bruce, C. C., Zhao, C. & Franklin, R. J. Remyelination — an effective means of neuroprotection. Horm. Behav. 57, 56–62 (2010).
Wang, J. et al. Activated regulatory T cell regulates neural stem cell proliferation in the subventricular zone of normal and ischemic mouse brain through interleukin 10. Front. Cell Neurosci. 9, 361 (2015).
Ishibashi, S. et al. Galectin-1 regulates neurogenesis in the subventricular zone and promotes functional recovery after stroke. Exp. Neurol. 207, 302–313 (2007).
Chan, A., Yan, J., Csurhes, P., Greer, J. & McCombe, P. Circulating brain derived neurotrophic factor (BDNF) and frequency of BDNF positive T cells in peripheral blood in human ischemic stroke: effect on outcome. J. Neuroimmunol. 286, 42–47 (2015).
Li, P. et al. Adoptive regulatory T-cell therapy protects against cerebral ischemia. Ann. Neurol. 74, 458–471 (2013).
Deliyanti, D. et al. Foxp3+ Tregs are recruited to the retina to repair pathological angiogenesis. Nat. Commun. 8, 748 (2017).
Leung, O. M. et al. Regulatory T cells promote apelin-mediated sprouting angiogenesis in type 2 diabetes. Cell Rep. 24, 1610–1626 (2018).
Cheng, Y. H. et al. Galectin-1 contributes to vascular remodeling and blood flow recovery after cerebral ischemia in mice. Transl. Stroke Res. 13, 160–170 (2022).
Lemaitre, P. et al. Molecular and cognitive signatures of ageing partially restored through synthetic delivery of IL2 to the brain. EMBO Mol. Med. 15, e16805 (2023).
Liston, A. & Yshii, L. T cells drive aging of the brain. Nat. Immunol. 24, 12–13 (2023).
Raffin, C., Vo, L. T. & Bluestone, J. A. Treg cell-based therapies: challenges and perspectives. Nat. Rev. Immunol. 20, 158–172 (2020).
Liston, A. & Gray, D. H. Homeostatic control of regulatory T cell diversity. Nat. Rev. Immunol. 14, 154–165 (2014).
Yamamoto, S. et al. In vitro generation of brain regulatory T cells by co-culturing with astrocytes. Front. Immunol. 13, 960036 (2022).
Chwojnicki, K. et al. Administration of CD4+CD25highCD127–FoxP3+ regulatory T cells for relapsing–remitting multiple sclerosis: a phase 1 study. BioDrugs 35, 47–60 (2021).
Kohm, A. P., Carpentier, P. A., Anger, H. A. & Miller, S. D. Cutting edge: CD4+CD25+ regulatory T cells suppress antigen-specific autoreactive immune responses and central nervous system inflammation during active experimental autoimmune encephalomyelitis. J. Immunol. 169, 4712–4716 (2002).
Maldini, C. R., Ellis, G. I. & Riley, J. L. CAR T cells for infection, autoimmunity and allotransplantation. Nat. Rev. Immunol. 18, 605–616 (2018).
Fransson, M. et al. CAR/FoxP3-engineered T regulatory cells target the CNS and suppress EAE upon intranasal delivery. J. Neuroinflammation 9, 112 (2012).
Kim, Y. C. et al. Engineered MBP-specific human Tregs ameliorate MOG-induced EAE through IL-2-triggered inhibition of effector T cells. J. Autoimmun. 92, 77–86 (2018).
Bittner, S., Hehlgans, T. & Feuerer, M. Engineered Treg cells as putative therapeutics against inflammatory diseases and beyond. Trends Immunol. 44, 468–483 (2023).
Pierson, W. et al. Antiapoptotic Mcl-1 is critical for the survival and niche-filling capacity of Foxp3+ regulatory T cells. Nat. Immunol. 14, 959–965 (2013).
Rosenzwajg, M. et al. Immunological and clinical effects of low-dose interleukin-2 across 11 autoimmune diseases in a single, open clinical trial. Ann. Rheum. Dis. 78, 209–217 (2019).
Louapre, C. et al. A randomized double-blind placebo-controlled trial of low-dose interleukin-2 in relapsing–remitting multiple sclerosis. J. Neurol. 270, 4403–4414 (2023).
Dansokho, C. et al. Regulatory T cells delay disease progression in Alzheimer-like pathology. Brain 139, 1237–1251 (2016).
Sheean, R. K. et al. Association of regulatory T-cell expansion with progression of amyotrophic lateral sclerosis: a study of humans and a transgenic mouse model. JAMA Neurol. 75, 681–689 (2018).
Guo, Z. et al. Low-dose interleukin-2 reverses chronic migraine-related sensitizations through peripheral interleukin-10 and transforming growth factor β1 signaling. Neurobiol. Pain. 12, 100096 (2022).
Huang, C., Zhang, F., Li, P. & Song, C. Low-dose IL-2 attenuated depression-like behaviors and pathological changes through restoring the balances between IL-6 and TGF-β and between TH17 and Treg in a chronic stress-induced mouse model of depression. Int. J. Mol. Sci. 23, 13856 (2022).
Thonhoff, J. R. et al. Combined regulatory T-lymphocyte and IL-2 treatment is safe, tolerable, and biologically active for 1 year in persons with amyotrophic lateral sclerosis. Neurol. Neuroimmunol. Neuroinflamm. 9, e200019 (2022).
Beers, D. R. et al. Tregs attenuate peripheral oxidative stress and acute phase proteins in ALS. Ann. Neurol. 92, 195–200 (2022).
Bensimon, G., Leigh, P. N. & Group, M. S. Modifying immune response and outcomes in ALS (MIROCALS): design and results of a phase 2b, double-blind randomized placebocontrolled trial of low dose interleukin-2 (ld IL2) in ALS. Platform Communications: Abstract Book—33rd International Symposium on ALS/MND, Amyotrophic Lateral Sclerosis and Frontotemporal Degeneration 23 (2022).
Ward, N. C. et al. IL-2/CD25: a long-acting fusion protein that promotes immune tolerance by selectively targeting the IL-2 receptor on regulatory T cells. J. Immunol. 201, 2579–2592 (2018).
Koten, J. W. et al. IL-2 loaded dextran microspheres with attractive histocompatibility properties for local IL-2 cancer therapy. Cytokine 24, 57–66 (2003).
Lopez, M. G., Diez, M., Alfonso, V. & Taboga, O. Biotechnological applications of occlusion bodies of Baculoviruses. Appl. Microbiol. Biotechnol. 102, 6765–6774 (2018).
de Picciotto, S. et al. Selective activation and expansion of regulatory T cells using lipid encapsulated mRNA encoding a long-acting IL-2 mutein. Nat. Commun. 13, 3866 (2022).
Teleanu, D. M., Chircov, C., Grumezescu, A. M., Volceanov, A. & Teleanu, R. I. Blood–brain delivery methods using nanotechnology. Pharmaceutics 10, 269 (2018).
Alves, S. et al. Interleukin-2 improves amyloid pathology, synaptic failure and memory in Alzheimer’s disease mice. Brain 140, 826–842 (2017).
Heimberger, A. B. et al. Incidence and prognostic impact of FoxP3+ regulatory T cells in human gliomas. Clin. Cancer Res. 14, 5166–5172 (2008).
Crane, C. A., Ahn, B. J., Han, S. J. & Parsa, A. T. Soluble factors secreted by glioblastoma cell lines facilitate recruitment, survival, and expansion of regulatory T cells: implications for immunotherapy. Neuro Oncol. 14, 584–595 (2012).
Han, S. et al. Tumour-infiltrating CD4+ and CD8+ lymphocytes as predictors of clinical outcome in glioma. Br. J. Cancer 110, 2560–2568 (2014).
van Hooren, L. et al. CD103+ regulatory T cells underlie resistance to radio-immunotherapy and impair CD8+ T cell activation in glioblastoma. Nat. Cancer 4, 665–681 (2023).
Amoozgar, Z. et al. Targeting treg cells with GITR activation alleviates resistance to immunotherapy in murine glioblastomas. Nat. Commun. 12, 2582 (2021).
Solomon, I. et al. CD25-Treg-depleting antibodies preserving IL-2 signaling on effector T cells enhance effector activation and antitumor immunity. Nat. Cancer 1, 1153–1166 (2020).
Author information
Authors and Affiliations
Contributions
The authors contributed equally to all aspects of the article.
Corresponding authors
Ethics declarations
Competing interests
The VIB and Babraham Institute are owners of patent applications PCT/GB2020/052148 and GB2118073.2, commented on in this work, with A.L., E.P. and L.Y. potential financial beneficiaries of commercialization. D.C.F. declares no competing interests.
Peer review
Peer review information
Nature Reviews Immunology thanks J. Bluestone and the other, anonymous, reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Liston, A., Pasciuto, E., Fitzgerald, D.C. et al. Brain regulatory T cells. Nat Rev Immunol 24, 326–337 (2024). https://doi.org/10.1038/s41577-023-00960-z
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41577-023-00960-z
