
Abstract
Background
Neurodevelopmental disorder with dysmorphic factors and distal skeletal anomalies (NEDDFSA) is a rare and phenotypically variable disorder. The zinc finger MIZ-type containing 1 gene (ZMIZ1) is a causative gene of NEDDFSA that encodes a protein inhibitor of the activated STAT-like family transcriptional regulator. Given the rarity of reported NEDDFSA cases, new phenotypes and genotypes of this disorder are still being discovered.
Objective
This study describes the phenotype characteristics of a Chinese NEDDFSA family caused by a novel ZMIZ1 variant.
Methods
We reviewed the clinical phenotype of a Chinese patient with NEDDFSA and performed whole-exome sequencing (WES) of the patient’s family. We simulated the potential biological harmfulness of the mutant protein. Plasmids were constructed and used for western blot and immunofluorescence assays to analyze protein expression levels.
Results
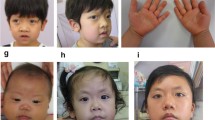
The patient was a 6-month-old male infant who exhibited dysmorphic facial features, neurodevelopmental abnormalities, congenital heart disease, and previously unreported genitourinary system anomalies. WES revealed a non-frameshift deletion variant in ZMIZ1 (NM_020338.4: c.858_875del, p.Val288_Ala293del), resulting in a structural alteration in the protein’s alanine-rich domain. Western blot and immunofluorescence assays indicated a significant decrease in the expression level of the mutant ZMIZ1 protein compared to the wild-type protein.
Conclusion
The clinical manifestations of this patient may be associated with the ZMIZ1 variant, and the structural alteration in the alanine-rich domain of the ZMIZ1 protein may contribute to a more complex disease phenotype. These results expand the genotype–phenotype correlation of ZMIZ1.




Similar content being viewed by others


Data Availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Beliakoff J, Lee J, Ueno H, Aiyer A, Weissman IL, Barsh GS, Cardiff RD, Sun Z (2007) The PIAS-like protein Zimp10 is essential for embryonic viability and proper vascular development. Mol Cell Biol 28:282–292
Carapito R, Ivanova EL, Morlon A, Meng L, Molitor A, Erdmann E, Kieffer B, Pichot A, Naegely L, Kolmer A et al (2019) ZMIZ1 variants cause a syndromic neurodevelopmental disorder. Am J Hum Genet 104:319–330
Chong S, Dugast-Darzacq C, Liu Z, Dong P, Dailey GM, Cattoglio C, Heckert A, Banala S, Lavis L, Darzacq X et al (2018) Imaging dynamic and selective low-complexity domain interactions that control gene transcription. Science 361:eaar2555
Latchman K, Calder M, Morel D, Rhodes L, Juusola J, Tekin M (2020) Autosomal dominant inheritance in a recently described ZMIZ1-related neurodevelopmental disorder: Case report of siblings and an affected parent. Am J Med Genet A 182:548–552
Lee J, Beliakoff J, Sun Z (2007) The novel PIAS-like protein hZimp10 is a transcriptional co-activator of the p53 Tumor suppressor. Nucleic Acids Res 35:4523–4534
Lu G, Ma L, Xu P, Xian B, Wu L, Ding J, He X, Xia H, Ding W, Yang Z et al (2022) A De Novo ZMIZ1 pathogenic variant for neurodevelopmental disorder with dysmorphic facies and distal skeletal anomalies. Front Genet 13:840577
Phetthong T, Khongkrapan A, Jinawath N, Seo GH, Wattanasirichaigoon D (2021) Compound heterozygote of Point Mutation and Chromosomal Microdeletion Involving OTUD6B coinciding with ZMIZ1 variant in Syndromic Intellectual disability. Genes (Basel) 12:1583
Pinnell N, Yan R, Cho HJ, Keeley T, Murai MJ, Liu Y, Alarcon AS, Qin J, Wang Q, Kuick R et al (2015) The PIAS-like Coactivator Zmiz1 is a direct and selective cofactor of Notch1 in T Cell Development and Leukemia. Immunity 43:870–883
Rakowski LA, Garagiola DD, Li CM, Decker M, Caruso S, Jones M, Kuick R, Cierpicki T, Maillard I, Chiang MY (2013) Convergence of the ZMIZ1 and NOTCH1 pathways at C-MYC in acute T lymphoblastic leukemias. Cancer Res 73:930–941
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E et al (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17:405–424
Rodriguez-Magadán H, Merino E, Schnabel D, Ramírez L, Lomelí H (2008) Spatial and temporal expression of Zimp7 and Zimp10 PIAS-like proteins in the developing mouse embryo. Gene Expr Patterns 8:206–213
Rodríguez-Magadán H, Ramírez L, Schnabel D, Vázquez M, Lomelí H (2010) Sexually dimorphic gene expression of the Zimp7 and Zimp10 genes in embryonic gonads. Gene Expr Patterns 10:16–23
Sharma M, Li X, Wang Y, Zarnegar M, Huang CY, Palvimo JJ, Lim B, Sun Z (2003) hZimp10 is an androgen receptor co-activator and forms a complex with SUMO-1 at replication foci. EMBO J 22:6101–6214
Shuai K, Liu B (2005) Regulation of gene-activation pathways by PIAS proteins in the immune system. Nat Rev Immunol 5:593–605
Yan B, Raben N, Plotz PH (2002) Hes-1, a known transcriptional repressor, acts as a transcriptional activator for the human acid alpha-glucosidase gene in human fibroblast cells. Biochem Biophys Res Commun 291:582–587
Zhou B, Lin W, Long Y, Yang Y, Zhang H, Wu K, Chu Q (2022) Notch signaling pathway: architecture, Disease, and therapeutics. Signal Transduct Target Ther 7:95
Acknowledgements
We are very grateful to the patient’s guardian for their understanding and support of this study.
Funding
This research was funded by the Anhui Province Key Research and Development Project (2022j11020003).
Author information
Authors and Affiliations
Contributions
LH, YW and LZ conceived and designed the study. LH and LZ contributed the sample collection and genotype data. LG checked partial data and language editing. LH, YW, JP and HZ interpreted the data and wrote the paper. All authors have read and approved the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that there is no conflict of interest regarding the publication of this paper.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
He, L., Wang, Y., Pan, J. et al. Clinical report and genetic analysis of a novel variant in ZMIZ1 causing neurodevelopmental disorder with dysmorphic factors and distal skeletal anomalies in a Chinese family. Genes Genom 46, 489–498 (2024). https://doi.org/10.1007/s13258-023-01480-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13258-023-01480-9