
Abstract
Mixtures containing isobutane, carbon dioxide, and/or hydrogen are found in various industrial processes, green refrigerant systems, and the growing hydrogen industry. Understanding the thermophysical properties of these mixtures is essential for these processes, and depends on reliable experimental data. Making use of an automated static-analytical apparatus, measurements were made of the phase behavior of binary mixtures of isobutane with CO2 and with H2, extending the range of available data for both mixtures. Measurements of the system isobutane + CO2 were carried out along three isotherms at temperatures of (240, 280, and 310) K with pressures from the lower limit of the sampling system (~ 0.5 MPa) to the mixture critical pressure. The results exhibit good agreement with literature data. Measurements on isobutane + H2 were carried out along nine isotherms at temperatures of (190, 240, 280, 311, 339, 363, 375, 390, and 400) K with pressures up to 20 MPa, covering a much broader range of conditions than the one prior investigation. The results have been used to optimize temperature-dependent binary parameters in the Peng–Robinson equation of state with two different mixing rules. This approach was found to perform well in comparison to alternative models.
Similar content being viewed by others


1 Introduction
Isobutane (2-methylpropane) is the simplest isomeric alkane, being a structural isomer of n-butane with a tertiary carbon atom at its center. The differences in its behaviour compared with n-butane can offer meaningful insights into how isomers behave, helpful both for a deeper understanding of chemical physics and the development of group contribution methods, especially for the functional group CH. Beyond theoretical value, isobutane is a substance of great importance in various existing and nascent processes, including those on the path to net zero.
An increasingly important application of isobutane is in the area of refrigeration, where, under the name R600a, it has become one of the leading “green” or “natural” refrigerants [1]. These are refrigerants with a low ozone depletion potential (ODP) and a low global warming potential (GWP). Isobutane has effectively zero ODP and a 100 year GWP of three [2]. It has a simple and stable structure, making it an ideal choice of refrigerant. Carbon dioxide (CO2, R744) is also a green refrigerant, with zero ODP and a GWP of one by definition. Blends of isobutane with CO2 have been considered and investigated [3,4,5,6,7] and offer a compromise on important properties, improving positive qualities like coefficient of performance while mitigating the negatives. For instance, isobutane is extremely flammable and so leaks present a fire risk, whereas CO2 is non-flammable and provides a reduction in mixture flammability. On the other hand, CO2 cycles operate at relatively high pressure and incur greater capital costs to withstand that pressure, while isobutane cycles operate at a much more moderate pressure. Beneficially, both are non-toxic, unlike ammonia, another potential green refrigerant [1]. Unfortunately, there is currently insufficient thermophysical property data available in the literature to develop refrigeration systems using isobutane + CO2 blends, in one case forcing propane + CO2 to be used as a substitute in modeling [3].
Isobutane is present in large scales across the energy and chemical industries. Isobutane is primarily produced from the isomerisation of n-butane [8], the majority of which is then used to produce 2,2,4-trimethylpentane, the fuel additive commonly known as isooctane, in alkylation units. It can also be produced from CO2 and hydrogen (H2) [9], either with a more classical route using syngas or a greener approach using captured CO2 and ‘blue’ or ‘green’ hydrogen. Knowledge of the behavior of isobutane with CO2 and H2 is thus highly pertinent to these processes as well as those downstream and ancillary, doubly so with regard to separations therein. These compounds will inevitably continue to persist as minor components and impurities far downstream of their introduction, including potentially in CO2 and H2 pipelines. Along a similar vein, research is being conducted into the blending hydrogen and natural gas, wherein isobutane is a relevant constituent under investigation [10, 11]. The critical temperatures and pressures of the pure compounds, along with their triple point temperatures, are listed in Table 1 for reference.
More generally, there is great and growing value in understanding the behavior of hydrogen mixtures, an area far less developed than for other industrial mixtures in juxtaposition to its importance in the ongoing energy transition. Even for common mixtures such as H2 with light alkanes, there is scant thermophysical property data available in the literature, much of which is antiquated [12,13,14,15,16] and lacking details of experimental uncertainty. This affects both existing and future processes, including H2 storage and transportation. Phase behavior and density data especially are needed as the key properties used to optimize and assess thermodynamic models.
There have been seven previous investigations into the phase behavior of isobutane + CO2 in the literature, as summarized in Table 2. The most recent study is that of Wang and Li [26], containing relatively few points with a substantial amount of scatter. The only other work that covered the low temperature region was Weber’s [22]; this data set comprises many points that are generally consistent, though there are a small number of apparently erroneous points visible, such as on the 280 K isotherm. A later study by Weber [24] covered higher temperatures, almost to the critical point of isobutane. The data here also appear to be of high quality, with little scatter visible except in the near critical region at T = 394 K. Three of the four isotherms were matched to those in the earliest study of isobutane + CO2, that of Besserer and Robinson [20], showing relatively close agreement though there are some minor disagreements for the vapor composition. One isotherm of Besserer and Robinson, at T = 378 K, exhibits some scatter, with two vapor points clearly out of line, but otherwise the data are satisfactory. Robinson, with Leu, later conducted another investigation of the phase behavior near to the critical point of isobutane [23], including measurements of the mixture critical point for the two temperatures. Nagahama et al. [21] conducted some measurements only at T = 273 K; however, like Wang and Li’s measurements at the same temperature, there is noticeable scatter and the data are not so consistent with those at different temperatures from other reliable studies. Finally, the second most contemporary work, that of Nagata et al. [25], reports data along four isotherms at temperatures between (300 and 330) K with only a minor degree of scatter.
There appears to have been only one prior investigation into isobutane + H2 reported in all of the open literature, for any thermophysical property. This is the work of Dean and Tooke [16], published in 1946, covering a temperature range from (311 to 394) K at six pressures from (3.45 to 20.69) MPa, from a series of studies of the phase behavior of H2 + hydrocarbons [13, 16, 27, 28]. The experiment used an apparatus first described in 1937 [29], state of the art for its day but with notable limitations. Two issues identified at the time were that the stirring system did not handle the non-lubricating nature of the fluids well, limiting stirring speeds and causing “extreme” equilibration times, and that it was difficult to prevent H2 leaking through the seal of a wire feedthrough. Additionally, the procedure employed to take samples and then analyze them, while advanced for the time, is rudimentary by modern standards. The convoluted method of separating the components involving freezing out isobutane, fractional distillation, and hydrogen scrubbing, followed by volume determination with balloons no doubt contribute significantly to the uncertainty. Mercury was also used to pressurize the system and this can influence the phase behavior, especially at higher temperatures. The limitations of this apparatus lead to the authors changing to an improved apparatus and an updated sampling technique for the two other mixtures investigated in that work.
2 Experimental Methods
2.1 Apparatus
The vapor-liquid equilibrium (VLE) apparatus used in this work, shown in Fig. 1, has been described in several previous publications [30,31,32], so not all details are described fully here, although modifications made since the previous publications are detailed. The technique employed was the static-analytic or analytical-isothermal method [33], where the vapor and liquid phases of a mixture in equilibrium at a known temperature and pressure are sampled and their compositions are quantitatively analyzed, in this case by gas chromatography (GC). The apparatus had a maximum working pressure of 20 MPa and could be operated in the temperature range of (183 to 473) K. This operating envelope permitted measurements over the full range of interest in this work, from the triple point of CO2 to the critical point of isobutane.

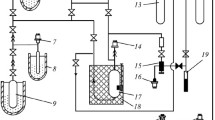
Simplified P&ID diagram of the VLE apparatus used in this work, adapted from [30] to include modifications made since previous publications
A high-pressure equilibrium cell contained the fluids under investigation. It was made of type 316L stainless steel and had an internal volume of approximately 143 cm3. The cell was sealed with a sliver-plated, nitrogen-pressurized hollow stainless steel O-ring. A blind hole in the wall of the cell housed a secondary-standard platinum resistance thermometer (Fluke model 5615), calibrated according to the International Temperature Scale of 1990 between (77 and 693) K and connected to a digital readout unit (Fluke 1502A). The resistance of this thermometer was checked immediately prior to this work in a triple-point-of-water cell. The overall estimated standard uncertainty in temperature was 0.006 K. Pressure measurements were made by the combination of a gauge pressure sensor (Keller PA-33X, 30 MPa full scale) and an absolute pressure sensor for the atmospheric pressure (Keller PAA-21Y, 0.2 MPa full scale), leading to an estimated standard uncertainty in pressure of 0.009 MPa. The cell was immersed in a thermostatic bath (Lauda Proline RP890). Three different bath fluids were used to cover the range of temperatures explored in this work. Ethanol was used for all of the isobutane + CO2 measurements and the low temperature isobutane + H2 measurements, deionised water was used for the measurements between (311 and 363) K, and silicone oil for the high temperatures. Mixing inside the cell was promoted by a polytetrafluoroethylene (PTFE)-coated magnetic stirrer bar magnetically coupled to an external permanent magnet and gear assembly driven by a motor above the bath.
The cell was filled and emptied using a fluid handling system, previously designed purely for gas handling but now adapted to allow for measurements of liquids and easily condensable gases. It featured a 500 cm3 reservoir equipped with a pressure sensor that allowed roughly known amounts of CO2 to be transferred to the cell, though in this work it was simply used as an intermediate store of CO2. Instead, knowledge of the cell contents relied on pVT relations and a newly added high-pressure syringe pump (Teledyne ISCO 100 DX) with electronic inlet and outlet valves. A fluid jacket around the syringe pump allowed for temperature control using a recirculating thermostatic bath (Huber CC1) filled with deionized water. This pump was connected to the cell via a 0.79 mm outer diameter (OD) × 0.50 mm internal diameter (ID) tube, which passed coaxially through the existing 3.18 mm OD inlet/outlet port and extended almost to the bottom of the cell. A second diaphragm vacuum pump (Vacuubrand MZ 2D NT) was connected upstream of the syringe pump to aid in evacuating the system. Toward the end of the investigation, a system to automatically vent the vapor phase was added. This consisted of a pneumatically operated inverted-bellows valve (Swagelok Nupro HB Series), actuated using a solenoid valve, downstream of a 2 m long × 0.13 mm ID capillary to help regulate the flow to a controllable rate. The fluid handling system also featured a nitrogen purge line to keep the bath blanketed with nitrogen.
Samples were withdrawn from the cell through two electromagnetic rapid on-line sample injector (ROLSI) valves (MINES-ParisTech ROLSI Evolution IV). These were connected to the cell with 1.59 mm OD × 0.13 mm ID capillaries, one passing near to the bottom of the cell, while the other stopped near the top, which allowed sampling from both the liquid and vapor phases. The sampling valves could be opened for durations from 10 ms to several seconds, allowing a sample, on the order of micromoles, to be drawn into the valve and swept into the GC by carrier gas. The small sample size relative to the cell volume minimized disturbances to the equilibrium. To avoid condensation, the valves were fitted with cartridge heaters and were insulated with glass-filled PTFE shells. These were maintained at T = 313 K for sub-ambient isotherms, to avoid temperature fluctuations, or generally 20 K above the cell temperature for super-ambient isotherms. The ROLSI valves were arranged in a parallel configuration and connected to the GC via a bespoke heated transfer line. A short section of the tubing immediately upstream of ROLSI valves and the entire sample return path were heated, with the sample facing surfaces passivated (SilcoTek Sulfinert) to the greatest extent possible. The GC (Agilent 7890A) allowed for the components to be separated on a packed column (Porapak Q) and quantified with a thermal conductivity detector (TCD). It also featured a gas calibration system described later.
The apparatus was controlled be a Keysight VEE program on a desktop computer, with a data acquisition unit (DAU) (Agilent 34970A) aiding data logging and switching. This program would start and stop the GC method and stirring system, actuate the ROLSI valves and venting valve, and interface with the syringe pump, allowing for a sequence of measurements to be run automatically. The venting system was operated on a closed loop with a simple on–off controller implemented in software.
2.2 Materials
The source and purity of the samples are described in Table 3. All were obtained from BOC and were used as supplied without further purification. Isobutane could only be obtained at a purity of 0.995; the main impurity is understood to be n-butane, whose properties are relatively close to those of isobutane.
Helium was used as the carrier gas for the isobutane + CO2 measurements, generally an ideal carrier gas when using a TCD because it has a much greater thermal conductivity than most other compounds, providing a strong response to both isobutane and CO2. Hydrogen is the only gas with a higher thermal conductivity than helium and the thermal conductivity of mixtures of hydrogen and helium does not vary as a monotonic function of mole fraction [34, 35]. Therefore, helium is not a suitable carrier gas when detecting hydrogen using a TCD and argon was used in its place for the isobutane + H2 measurements. The thermal conductivity of argon is much lower than that of hydrogen and approximately half that of isobutane, meaning that a strong response was obtained for the former, while an acceptable response was obtained for the latter.
2.3 Calibration
Calibration of the GC system was performed in the manner described previously, employing the absolute-area method, the accuracy and effectiveness of which is well validated [30,31,32]. Here, the detector response to sample analytes is related to that produced by known amounts of pure analyte. The GC was equipped with two gas sampling loops, of nominal volume 0.05 cm3 and 0.2 cm3. Each loop had two K-type thermocouples affixed to it, yielding a standard uncertainty in temperature of 1 K, and an in-line pressure sensor (Keller PAA-21Y, 0.6 MPa full scale), with relative standard uncertainty of 0.3%. These loops could be filled with vapor samples of the pure components being investigated, the densities of which were calculated with low relative uncertainty based on the measured temperature and pressure. The equations of state (EoS) [17,18,19] used, as implemented in REFPROP 10.0 [36], to calculate the densities are listed in Table 4 along with their stated relative expanded uncertainties (k = 2). The contents of these loops are injected into the GC upstream of the column and produce a chromatographic peak upon passing through the detector. A calibration curve may be built up by injecting samples across a range of different loop filling pressures, from (0.1 to 0.3) MPa.
The loop filling density of component \(i\), \({\rho }_{i,{\text{loop}}}\), and by extension the amount, \({n}_{i}\), were described as a quadratic function of the peak area as follows:
where \({V}_{{\text{loop}}}\) is the volume of the gas sampling loop, \({A}_{i}\) is the peak area produced by the TCD by component \(i\), and \({a}_{1,i}\) and \({a}_{2,i}\) are substance-specific calibration constants regressed by minimisation of the sum-squared error \(S\)
between the filling density and the density predicted by the quadratic equation, \({\rho }_{i,{\text{calc}}}\). A quadratic relationship was used to capture the effect of slightly non-linear detector responses, not uncommon for TCDs [37], and was found to perform notably better than the linear form. The optimized calibration constants for each loop are listed in Table 5. These constants are specific to a given loop volume; however, in practice, the amount of a sample is immaterial and only the mole fraction is relevant, in the calculation of which the volumes cancel out. As two loops of different volumes were used an average is taken of the mole fractions predicted from either set of constants.
2.4 Procedure
The procedure began with flushing the system (VLE cell, syringe pump, tubing, etc.) with the experimental gases before evacuating using the two vacuum pumps. The syringe pump was chilled to T = 278 K and isobutane allowed to flow in from the cylinder, condensing within and filling the pump. The pump temperature was then increased to T = 293 K to avoid water condensing on its exterior while still maintaining a constant temperature. Once the temperature had equilibrated, isobutane was injected into the VLE cell.
In the absence of a window for visual confirmation, the volume had to be determined in advance to ensure that the ultimate liquid level inside the cell would lie between the two capillaries, such that they could draw appropriate samples. This required knowledge of the pVT conditions of the pump and the VLE cell and the ability to model the mixture behavior approximately. In this case, the Peng–Robinson (PR) EoS detailed in Sect. 4.1 was used. Both the pump and cell were held at fixed (though generally unequal) temperatures, and the cell was of fixed, approximately known, volume. The density of isobutane at the pump conditions was predicted by the model, providing knowledge of the amount of isobutane entering the cell for a given change in pump volume. The compositions of the vapor and liquid phases, and their densities, at the desired experimental pressure were also be predicted by the model, resulting in a sufficiently described system to determine the liquid level.
With the isobutane injected, the pressure in the cell was raised to the desired value, using either CO2 or H2 from a cylinder, and allowed to equilibrate while stirring. In cases where the desired pressure was greater than could be delivered from the cylinder, higher pressures could be achieved either by injecting more isobutane from the pump or by filling the cell at a lower temperature and then carefully allowing the contents of the cell to heat up isochorically.
The stirrer was stopped ahead of sampling, allowing the liquid to settle and preventing any vortex potentially wetting capillaries with other phase or foaming issues arising. A quick series of liquid samples would be taken to flush the capillary, followed by five to ten liquid samples to be used in analysis, at five minute intervals. Samples of the vapor phase were then taken, up to twenty five at ten minute intervals. In some cases, as many as fifty vapor samples were taken in order to observe the effect of further sampling on sample composition. Initially a period of vapor capillary flushing was used; however, it was found to be able to disturb the equilibrium too greatly and was omitted in later measurements. Afterward, stirring was resumed and one could move to next pressure point, either by manually venting some of the vapor phase to lower pressure or by injecting components to raise pressure.
2.5 Automation
Automated measurements of a narrow range of pressures can be achieved with only one degree of freedom, this was achieved by injecting isobutane from the ISCO pump, raising the pressure while also increasing the overall mole fraction of isobutane. A consequence of this method is that the liquid level inside the cell rises quickly as pressure increases, eventually immersing the vapor capillary followed by the system leaving the two phase region. This limits the range of pressures that can be accessed during a single run, especially when starting at lower pressures.
Automated measurements of entire isotherms, for the mixtures investigated, require the ability to move through the pxy space with two degrees of freedom, varying both pressure and the overall composition. This was achieved through a combination of venting the vapor phase and injecting isobutane from the pump. Venting the vapor phase reduces the pressure rather strongly while decreasing the overall mole fraction of the more volatile component (CO2 or H2). As this happens, the liquid level inside the cell would decrease, which could be counteracted by injecting isobutane from the pump, increasing the pressure slightly while increasing the isobutane mole fraction quickly.
3 Uncertainty
The uncertainty analysis in this work follows the same methodology as previous publications [30,31,32, 38] guided by the ‘Evaluation of Measurement Data–Guide to the Expression of Uncertainty in Measurement” recommended by NIST [39]. For the determination of the composition, \({z}_{i}\), of the liquid, \({x}_{i}\), or vapor, \({y}_{i}\), phases, the measured quantities were the temperature, \(T\), pressure, \(p\), and the chromatographic peak areas, \({A}_{i}\). The combined standard uncertainty in mole fraction \({u}_{c}\left({z}_{i}\right)\) is calculated as follows:
where \(u\left({z}_{i}\right)\) is the uncertainty in mole fraction, \(u\left(T\right)\) is the uncertainty in temperature, and \(u\left(p\right)\) is the uncertainty in pressure. The uncertainty in mole fraction is the main contribution in most cases, except where the mole fraction changes quickly such as near the critical point, and is calculated as follows:
where \({u}_{r}\left({A}_{i}\right)\) is the relative standard uncertainty in peak area and \({u}_{r}\left({f}_{i}\right)\) is the relative standard uncertainty in response factor, \({f}_{i}\). The latter comes from the calibration measurements and is given by:
where \({u}_{r}\left({n}_{i}\right)\) is the relative standard uncertainty in the amount of substance, \({n}_{i}\). As previously, \(u\left({z}_{i}\right)\) reduces to the simple quadratic expression:
The uncertainties in temperature and pressure were 0.006 K and 0.009 MPa, as mentioned above. The partial derivatives \(\left(\partial {z}_{i}/\partial T\right)\) and \(\left(\partial {z}_{i}/\partial p\right)\) were determined using the optimized EoS models, except for the near critical region for the (390 and 400) K isotherms in the isobutane + H2 measurements, where the scaling correlations described in Sect. 5.3 were used instead.
4 Modeling Methods
4.1 Peng–Robinson
Thermodynamic modeling has been carried out primarily using two variants of the Peng–Robinson EoS [40], computed by a VBA program implemented in Microsoft Excel. Both used the volume translation of Péneloux [41] and the Soave alpha function [42], but differed in mixing rules. One used the classical van der Waals mixing rules (PR-vdW), while the other used the modified Wong-Sandler mixing rules of Orbey and Sandler coupled with the NRTL free energy model (PR-mWS-NRTL) [43]; the latter can perform better for highly non-ideal mixtures. The significant equations within these model variants are explained below, the rest are excluded for brevity but can be found in the literature [43] along with discussion comparing the modified Wong–Sandler rule to the original.
For a mixture of \(n\) components, the main equation of the PR EoS is:
where \(p\) is the pressure, \(R\) the gas constant, \(T\) the temperature, \({V}_{{\text{m}}}\) the molar volume, and \({a}_{{\text{m}}}\) and \({b}_{{\text{m}}}\) are parameters calculated from the mixing rules. The van der Waals mixing rules are:
where \({x}_{i}\) and \({x}_{j}\) are the mole factions of components \(i\) and \(j\), respectively, \({a}_{i}\) and \({b}_{i}\) are substance-specific parameters, and \({k}_{ij}\) and \({l}_{ij}\) are the binary interaction parameters.
While the modified Wong-Sandler mixing rules are:
where \(Q\) and \(D\) are calculated as follows:
where \({x}_{i}\) and \({x}_{j}\) are the mole factions of components \(i\) and \(j\), respectively, \({\left(b-\frac{a}{RT}\right)}_{ij}\) is the second virial coefficient, \({a}_{i}\), \({\alpha }_{i}\), and \({b}_{i}\) are substance-specific parameters, \(C\) is a constant, and \({A}_{\infty }^{E}\) is the excess Helmholtz free energy at infinite pressure calculated from the NRTL free energy model. Contained within the equations for the second virial coefficient and the excess Helmholtz free energy at infinite pressure are the binary interaction parameters \({k}_{ij}\), \({\tau }_{ij}\), and \({\alpha }_{ij}\), specific to a given mixture, the last of which is kept constant (\({\alpha }_{ij}\) = 0.3), while the former two must be regressed against experimental data.
The binary interaction parameters used in both mixing rules were described as functions of temperature as follows:
where \(X\) stands for \({l}_{ij}\), \({\tau }_{12}\), or \({\tau }_{21}\), and \({c}_{1}\), \({c}_{2}\), and \({c}_{3}\) are constants regressed by minimizing the objective function \(S\):
where \(N\) is the number of state points included in the regression, \({x}_{1,i}\) is the mole fraction of component 1 at point \(i\) from experimental measurements, and \({x}_{1,i,\tt calc}\) is the mole fraction at that point calculated by the model. Component 1 denotes the more volatile component, CO2 or H2, while component 2 denotes the less volatile component, isobutane.
The model requires pure component critical parameters and acentric factors as inputs. As discussed in a previous publication [30], there are complications with this for hydrogen due to its quantum behavior [44]. As such, adjusted values were used to allow for better predictions at temperatures above the true critical temperature of hydrogen, determined by minimizing the error in the predicted second and third virial coefficients for pure hydrogen. As previously, the critical temperature was set to 31.76 K, the critical pressure to 1.276 MPa, and the acentric factor to − 0.0626.
4.2 Other Equations of State
The phase behavior has also been modeled with other types of equation of state to provide a comparison and demonstration of the effectiveness of the PR models, which use experimentally regressed parameters, while the other models are limited to predictive use. These models were used as implemented in the Julia software Clapeyron [45], whose wide selection of models and open source, thus verifiable, nature is most convenient. Numerous models could be examined, but this shall be limited to only a few, considered to be the most relevant.
One such model is a predictive version of PR, the Enhanced Predictive Peng Robinson (EPPR78) EoS [46, 47], which take a group contribution approach to determining BIPs, making it very versatile and suitable for use with mixtures that lack experimental data. The quality of the predictions is strongly dependent on the description of the functional groups and their interactions, so high-quality experimental data for similar, especially simpler, compounds are invaluable. The CH group, present in the center of an isobutane molecule, is poorly developed relative to the more common groups, such as CH3, three of which are present in isobutane. Fortunately, for EPPR78, the group interaction parameters are present for the CH group with CO2 and with H2. Another group contribution EoS is SAFT-γ-Mie [48], based on the statistical associating fluid theory (SAFT) using Mie potentials to describe group interactions. It is significantly more sophisticated than the older, cubic EoS, but is not as thoroughly developed, lacking the necessary parameters to model isobutane + H2, limiting its use to isobutane + CO2. It may be able to predict caloric properties more accurately [49], which are important for modeling refrigeration processes. The final model examined is GERG-2008 [50], a highly sophisticated Helmholtz energy explicit multiparameter equation of state developed for natural gases and similar mixtures, optimized for 21 compounds, including those in this work.
5 Results and Discussion
5.1 Isobutane + CO2
The phase behavior of isobutane + CO2 was investigated at three temperatures: (240, 280, and 310) K, with data tabulated in Table 6 and is shown graphically in Fig. 2. There were three literature data sources available around 310 K, one at 310 K [25] and two at 310.93 K [20, 24], and one source of literature data available at 280 K [22], allowing for comparisons. The data obtained in this work for those two isotherms were found to be in very good agreement with the literature data, providing helpful validation for the apparatus and technique, previously validated before the aforementioned modifications were made. It can be seen that the vapor composition at 3.34 MPa from Weber’s 280 K [22] measurements is probably erroneous, though the other measurements were of high quality. The 240 K isotherm is 10 K lower than in the next lowest temperature source [22], extending the available data further into the range of relevance for refrigeration. At 240 K, the pressures were nearing the lower limit of pressure at which the sampling system could function and required long ROLSI valve opening times, up to 10 s for the vapor phase, also meaning no measurements could be made at lower still temperatures. Even at these low pressures, the results appear consistent with the model predictions and higher temperature data.

pxy diagrams for [CO2 (1) + isobutane (2)] at (a) 310 K, (b), 280 K, and (c) 240 K, showing the experimental data from this work ●, as well as the data of Nagata et al. [25] at 310 K ○, Besserer and Robinson [20] at 310.93 K ◊, Weber [22, 24] at 310.93 K □ and 280 K. Also shown are the predictions of PR-vdW  and PR-mWS-NRTL
and PR-mWS-NRTL  and those of EPPR78
and those of EPPR78  , SAFT-γ-Mie
, SAFT-γ-Mie  , and GERG-2008
, and GERG-2008  (Color figure online)
(Color figure online)
Both PR-vdW and PR-mWS-NRTL were found to describe the behavior of isobutane + CO2 very closely, the accuracy primarily only limited by the scatter of the literature data (Table 7). A marginally better fit could be obtained for PR-mWS-NRTL, with an average absolute deviation in mole fraction of 0.0071 compared to 0.0094; however, the simpler PR-vdW would be sufficient in most cases. The former may predict the liquid phase density more accurately [43]; however, this was not able to be checked at this stage and is the subject of further work.
Comparisons of the predictions of the PR models with those of EPPR78, SAFT-γ-Mie, and GERG-2008 are also seen in Fig. 2. At temperatures of (280 and 310) K, the predictions of EPPR78 and GERG-2008 are relatively similar to each other and are almost in agreement with the experimental data; however, the bubble pressure is consistently underpredicted. At T = 240 K, the differences between them are more pronounced, with EPPR78 overpredicting the bubble pressure while GERG-2008 underpredicts it. The vapor compositions for these three isotherms are in close agreement between the PR models, EPPR78, and GERG-2008, though PR-mWS-NRTL just outperforms the others, more so with the liquid compositions. SAFT-γ-Mie has the most marked difference in its predictions. At T = 310 K, the predicted compositions are more isobutane-rich, especially for the liquid phase, and the critical pressure is slightly underpredicted. At T = 280 K, an unphysical azeotrope appears, also predicted at T = 240 K, and the liquid phase especially remains overly isobutane-rich with bubble pressures significantly overpredicted. A prediction of vapor–liquid–liquid equilibrium (VLLE) is also visible, which was not found to occur during experiments. If a three phase line were to exist, it is likely that this would be at temperatures lower than those at which solid formation, not normally considered in fluid thermodynamic models, occurs. As such, from a practical perspective, one could consider the mixture to exhibit type I phase behavior according to the classifications of Scott and van Konynenburg [51], as has been done for other CO2 + alkane mixtures [52]. For n-alkanes, it is only for heptane and beyond that the VLLE curve appears, so this treatment may be reasonable. As the Scott–van Konynenburg types do not consider solid formation, instead only interaction parameters, from a fluid theory perspective, one could perhaps view the mixture differently. Determining which type a mixture should be simply from interaction parameters is difficult for real mixtures, and the idealized analysis of Scott and van Konynenburg [51] suggests that the mixture should be classified as type II, but it is unclear if there is a physically meaningful difference between a type I mixture and a type II mixture where solid formation precedes a hypothetical liquid–liquid separation.
5.2 Isobutane + H2
The phase behavior of isobutane + H2 was investigated at nine temperatures: (190, 240, 280, 311, 339, 363, 390, and 400) K, with data tabulated in Table 8 and is shown graphically in Fig. 3. These measurements span almost the entire range from the lower temperatures limit of the apparatus to the critical point of isobutane, 71% of the range between the triple point and critical point of isobutane. For the (390 and 400) K isotherms, the mixture critical pressure was within the operating range of the apparatus, allowing for measurements to be made in the near critical region. This is very important for modeling purposes, as accuracy in this region can provide insights into a model’s performance and allow for better fitting. Predictions of this region were found to be much more variable when regressing BIPs only from the lower temperature isotherms, where the liquid composition varies relatively linearly as a function of pressure, akin to the idealistic Henry’s law, and the top of the phase envelope is far out of sight. Two isotherms, (311 and 339) K, could be directly compared to literature data [16], finding surprisingly close agreement for the liquid compositions, while there is a pronounced difference for the vapor compositions, with consistently lower hydrogen mole fractions found in this work than in literature. This is not unusual, given that the one prior study was conducted long ago with rudimentary techniques.


pxy diagrams for [H2 (1) + isobutane (2)] at (a) 190 K, (b), 240 K, (c) 280 K, (d) 311 K, (e) 339 K, (f) 363 K, (g) 375 K, (h) 390 K, and (i) 400 K, showing the experimental data from this work ●, as well as the data of Dean and Tooke [16] at (311 and 339) K ○ and the predictions of PR-vdW  (Color figure online)
(Color figure online)
There were some complications with measurements, most notably issues with sampling the vapor phase. Liquid samples were generally unproblematic, near identical samples of the liquid phase could be obtained immediately after a short flushing, while equivalently consistent samples of the vapor frequently required up to twenty five samplings, sometimes still varying unsatisfactorily even after that point. It is believed that these issues were the result of disruptions to the cell equilibrium upon sampling, where small amounts of liquid could become entrained in the vapor and even accumulate on the capillary for the vapor sampling ROLSI valve. These effects, and the extent of their inconvenience, have been detailed in some depth in the literature [53, 54] where windowed cells have allowed for visual observation. In some instances, such as some moderate pressures at 390 K, liquid compositions are given, while vapor compositions are not, as no steady vapor sample composition could be obtained despite the liquid samples remaining well behaved. A small number of state points are missing a liquid or vapor composition due to complications with liquid level. While implementing and testing the automated system, the liquid level inside the cell would sometimes exceed the working limits, especially when only using the syringe pump to increase the pressure before having the venting system in place. This was amplified by uncertainties from the model affecting estimates of the liquid level. This issue was normally exhibited as the first and/or last state point in a sequence sampling the same phase with both ROLSIs. These data were included when there was confidence that the contents of the cell were still in the two phase region, and excluded wherever it was likely that only one phase was present.
A quality of fit like that seen with isobutane + CO2 using the PR models could not be achieved with isobutane + H2. For PR-vdW, predictions of the vapor phase compositions are generally accurate; however, at higher temperatures, most notably (363 and 375) K, a difference in trend is visible at high pressures. The accuracy of the predicted liquid phase compositions varies less consistently across the temperature range, overpredicting the H2 mole fractions at (390 and 400) K, under-predicting them from (280 to 363) K, then notably overpredicting them again at 190 K. This is believed to be an artifact of the temperature-dependent BIPs (Table 9), especially given the good agreement between this work and the literature for the comparable isotherms. Unfortunately, numerical problems were encountered when trying to regress the BIPs for PR-mWS-NRTL for isobutane + H2, and no useful results could be obtained with this model. The highly asymmetric nature of this mixture greatly increases the difficulty in converging the flash algorithms, resulting in frequent, though unpredictable, failures to converge to a valid solution.
Despite the challenges in modeling isobutane + H2 with PR-vdW and PR-mWS-NRTL, a significantly better fit is obtained with PR-vdW than with the alternative models, as seen in Fig. 4. As mentioned previously, SAFT-γ-Mie could not be used for this mixture; however, EPPR78 and GERG-2008 could be. At T = 390 K, EPPR78 systematically underpredicts the isobutane mole fraction in both phases while overpredicting the critical pressure. While imprecise, the predictions are still reasonable and the trends as expected, the same statement cannot be made about the predictions of GERG-2008. At T = 390 K, the predicted phase envelope is much larger than found in experiments, with the liquid excessively isobutane-rich and the vapor excessively H2-rich, and does not close at high pressures. The shortcomings are most visible when examining the pressure–temperature plot of the mixture critical loci, as shown in Fig. 5. The experimentally estimated critical points and those predicted by PR-vdW and EPPR78 all depart from the critical point of isobutane with a negative slope and tend quickly toward great pressure, while GERG-2008 predicts that the critical loci depart with a positive slope before passing a temperature maximum and turning back to lower temperatures and higher pressures. This prediction is unphysical, the departure with positive slope is seen almost exclusively with helium mixtures [55], while the temperature maximum is more unusual still. Neither EPPR78 nor GERG-2008 produces sufficiently accurate predictions for real-world use, and there is a clear room for their parameters to be refined. Additionally, the group interaction parameters for the CH group with H2 in SAFT-γ-Mie can and should be optimized with the new experimental data, though this is outside the scope of this work.

pxy diagram for [H2 (1) + isobutane (2)] at 390 K, showing the experimental data from this work ●, as well as the predictions of PR-vdW  and those of EPPR78
and those of EPPR78  and GERG-2008
and GERG-2008  (Color figure online)
(Color figure online)

pT diagram for isobutane + H2, showing the vapor curves of pure isobutane –– and H2 predicted by the EoS of Bücker and Wagner and of Leachman et al., the critical points of pure isobutane ◆ and H2
predicted by the EoS of Bücker and Wagner and of Leachman et al., the critical points of pure isobutane ◆ and H2 , the experimentally estimated mixture critical points ●, and the mixture critical loci predicted by PR-vdW
, the experimentally estimated mixture critical points ●, and the mixture critical loci predicted by PR-vdW  , EPPR78
, EPPR78  , and GERG-2008
, and GERG-2008  (Color figure online)
(Color figure online)
The binary mixture of isobutane + H2 was found to unambiguously display type III [51] phase behavior, as could reasonably be expected for a mixture of such dissimilar molecules [55]. The mixture critical loci can be seen to rise sharply from the critical point of isobutane, though its path at very high pressures is unknown. The melting line of isobutane, near to which the three phase vapor-liquid–solid equilibrium line is expected to follow if consistent with other hydrogen mixtures [56,57,58,59], is far separated from the critical loci. It is perhaps unlikely that the two should ever meet, if so it would be at enormous pressure; however, the critical loci may instead eventually turn back toward higher temperatures, as is known to occur for other type III mixtures [55, 60]. Prediction of the phase behavior in regions so detached from experimental data is challenging, and the cubic equations of state used in this work may not be capable of describing some aspects of it [61], though alternative models such as Carnahan–Starling-type equations of state may overcome this issue.
5.3 Isobutane + H2 Critical Point Estimation
Critical scaling laws were applied in order to determine the critical point at (390 and 400) K, given in Table 10, using the following equation [62] for binary mixtures:
where \({z}_{1}\) is the mole fraction of H2 in the liquid, where \(\varepsilon =1\), or vapor, where \(\varepsilon =-1\), \(p\) is the pressure, \({z}_{1,{\text{c}}}\) is the critical composition, \({p}_{\text c}\) is the critical pressure, \(\beta =0.325\), and \({\lambda }_{1}\), \({\lambda }_{2}\), and \(\mu\) are scaling parameters (Fig. 6). \({z}_{1,{\text{c}}}\), \({p}_{\text c}\), \({\lambda }_{1}\), \({\lambda }_{2}\), and \(\mu\) have been described as a linear function of temperature, in order to allow for the calculation of \(\left(\partial {z}_{i}/\partial T\right)\) in Eq. 3, according to the equation:
where \(M\) stands for \({z}_{1,{\text{c}}}\), \({p}_{\text c}\), \({\lambda }_{1}\), \({\lambda }_{2}\), or \(\mu\), while \({a}_{1}\) and \({a}_{2}\) are constants are regressed against experimental data using the same objective function given in Eq. 16, and the values of which are listed in Table 11.

Near critical region of [H2 (1) + isobutane (2)] at (390 and 400) K in terms of the pressure \(p\) and reduced critical composition \({z}_{1}/{z}_{1,{\text{c}}}\), showing the predictions of the scaling law — as well as the experimental data for the liquid ○ and vapor ● compositions
The points at 9.18 MPa for the 400 K isotherm were initially excluded from the parameter fitting as both ROLSI valves had sampled the same phase and it was not certain if the system was still in the two phase region. These points were found to lie almost exactly on the curve predicted by the equations and so were ultimately included in the fitting, to minimal effect.
6 Conclusions
Mixtures containing isobutane, CO2, and H2 are relevant to current and future processes, but it was found that insufficient data were available in the literature for effective process development. To address this, state of the art measurements were made of the phase behavior of isobutane with CO2 and with H2, three isotherms for the former and nine for the latter, extending the range of available data for both mixtures. Good agreement with the literature was found for isobutane + CO2, which has provided validation for the updated and increasingly automated apparatus. The new measurements of the system isobutane + H2 represent the vast majority of thermophysical property measurements ever made of this mixture. A marked difference in vapor compositions compared to the one prior study was observed. Measurements in the near critical region could be made for the two highest temperature isotherms, and could be well described by critical scaling laws. The binary interaction parameters for two variants of the Peng Robinson equation of state, PR-vdW and PR-mWS-NRTL, have been optimized as functions of temperature. A quadratic function was most effective for \({k}_{ij}\), while linear functions were sufficient for the parameters \({l}_{ij}\) and \({\tau }_{ij}\). These models performed exceptionally well for isobutane + CO2 across the entire temperature range with available data, with PR-mWS-NRTL achieving a marginally better fit. For isobutane + H2, PR-vdW could produce a moderately good fit of the data, while we were unable to reliably converge flash calculations for PR-mWS-NRTL. In all cases, the optimized PR models outperformed the predictive group contribution models, EPPR78 and SAFT-γ-Mie, and the sophisticated multiparameter GERG-2008. The data generated also provide an avenue to refining interaction parameters for the CH functional group.
Data Availability
All relevant data are tabulated in the paper. No datasets were generated or analysed during the current study.
References
V.W. Bhatkar, V.M. Kriplani, G.K. Awari, Int. J. Environ. Sci. Technol. 10, 871-880 (2013)
IPCC, AR4, WG1, pp 210–216 (2007)
J. Sarkar, S. Bhattacharyya, Int. J. Therm. Sci. 48, 1460-1465 (2009)
P. Ganesan, T.M. Eikevik, K. Hamid, R. Wang, H. Yan, Int. J. Refrig. 154, 215-230 (2023)
X. Zhang, A. Chen, H. Duan, 2010 International Conference on Mechanic Automation and Control Engineering, Wuhan (2010)
X. Fan, F. Ju, X. Zhang, F. Wang, Therm. Sci. 18, 1655-1659 (2014)
M. Sobieraj, M. Rosiński, Int. J. Refrig. 103, 243-252 (2019)
Y. Ono, Catal. Today 81, 3-16 (2003)
H. Wang, S. Fan, S. Guo, S. Wang, Z. Qin, M. Dong, H. Zhu, W. Fan, J. Wang, Nat. Commun. 14, 2627 (2023)
EMPIR, Decarb, 20IND10 (2021)
R. Romeo, G. Cavuoto, P.A.G. Albo, S. Lago, ECTP 2023, Venice (2023)
D.B. Trust, F. Kurata, AIChE J. 17, 86-91 (1971)
H.J. Aroyan, D.L. Katz, Ind. Eng. Chem. 43, 185-189 (1951)
E.E. Nelson, W.S. Bonnell, Ind. Eng. Chem. 35, 204-206 (1943)
A.E. Klink, H.Y. Cheh, E.H. Amick, AIChE J. 21, 1142-1148 (1975)
M.R. Dean, J.W. Tooke, Ind. Eng. Chem. 38, 389-393 (1946)
D. Bücker, W. Wagner, J. Phys. Chem. Ref. Data 35, 929-1019 (2006)
R. Span, W. Wagner, J. Phys. Chem. Ref. Data 25, 1509-1596 (1996)
J.W. Leachman, R.T. Jacobsen, S.G. Penoncello, E.W. Lemmon, J. Phys. Chem. Ref. Data 38, 721-748 (2009)
G.J. Besserer, D.B. Robinson, J. Chem. Eng. Data 18, 298-301 (1973)
K. Nagahama, H. Konishi, D. Hoshino, M. Hirata, J. Chem. Eng. Jpn. 7, 323-328 (1974)
L.A. Weber, Cryogenics 25, 338-342 (1985)
A.D. Leu, D.B. Robinson, J. Chem. Eng. Data 32, 444-447 (1987)
L.A. Weber, J. Chem. Eng. Data 34, 171-175 (1989)
Y. Nagata, K. Mizutani, H. Miyamoto, J. Chem. Thermodyn. 43, 244-247 (2011)
H. Wang, D. Li, Shiyou-Haugong 46, 56-61 (2017)
A.L. Benham, D.L. Katz, AIChE J. 3, 33-36 (1957)
A.L. Benham, D.L. Katz, R.B. Williams, AIChE J. 3, 236-241 (1957)
D.L. Katz, K.H. Hachmuth, Ind. Eng. Chem. 29, 1072–1077 (1937)
O. Fandiño, J.P.M. Trusler, D. Vega-Maza, Int. J. Greenh. Gas Control 36, 78-92 (2015)
L.F.S. Souza, S.Z.S. Al-Ghafri, J.P.M. Trusler, J. Chem. Thermodyn. 126, 63–73 (2018)
L.F.S. Souza, S.Z.S. Al-Ghafri, O. Fandiño, J.P.M. Trusler, Fluid Ph. Equilib. 522, 112762 (2020)
R. Dohrn, S. Peper, J.M.S. Fonseca, Fluid Phase Equilib. 288, 1-54 (2010)
R.S. Hansen, R.R. Frost, J.A. Murphy, J. Phys. Chem. 68, 2028-2029 (1964)
A.G. Shashkov, F.P. Kamchatov, T.N. Abramenko, J. Eng. Phys. 24, 461-464 (1973)
E.W. Lemmon, I.H. Bell, M.L. Huber, M.O. McLinden, REFPROP 10.0, NIST (2018)
P.L. Patterson, R.A. Gatten, J. Kolar, C. Ontiveros, J. Chromatogr. Sci. 20, 27-32 (1982)
S.Z.S. Al-Ghafri, E. Forte, G.C. Maitland, J.J. Rodriguez-Henriquez, J.P.M. Trusler, J. Phys. Chem. B 118, 14461–14478 (2014)
BIPM, JCGM 100:2008 (2010)
D.Y. Peng, D.B. Robinson, Ind. Eng. Chem. Fundam. 15, 59-64 (1976)
A. Péneloux, E. Rauzy, R. Fréze, Fluid Phase Equilib. 8, 7-23 (1982)
G. Soave, Chem. Eng. Sci. 27, 1197-1203 (1972)
H. Orbey, S.I. Sandler, Cambridge Series in Chemical Engineering, (1998)
R.D. Gun, P.L. Chueh, J.M. Prausnitz, AIChE J. 12, 937-941 (1966)
P.J. Walker, H. Yew, A. Riedemann, Ind. Eng. Chem. Res. 61, 7130-7153 (2022)
J.N. Jaubert, R. Privat, F. Mutelet, AIChE J. 56, 3225-3235 (2010)
J.N. Jaubert, J.W. Qian, S. Lasala, R. Privat, Fluid Ph. Equilib. 5, 113456 (2022)
V. Papaioannou, T. Lafitte, C. Avendaño, C.S. Adjiman, G. Jackson, E.A. Müller, A. Galindo, J. Chem. Phys. 140, 054107 (2014)
A.J. Haslam, private communications (2023)
O. Kunz, W. Wagner, J. Chem. Eng. Data 57, 3032-3091 (2012)
P.H. van Konynenburg, R.L. Scott, Philos. Trans. R. Soc. Lond. 298, 495-540 (1980)
D.J. Fall, K.D. Luks, J. Chem. Eng. Data 30, 276-279 (1985)
F.C.V.N. Fourie, C.E. Schwarz, J.H. Knoetze, Chem. Eng. Technol. 38, 1165–1172 (2015)
F.C.V.N. Fourie, C.E. Schwarz, J.H. Knoetze, Chem. Eng. Technol. 39, 1475–1482 (2016)
J.S. Rowlinson, F.L. Swinton, Butterworths Monogr. Chem. 15, 191–229 (1982)
C.Y. Tsang, P. Clancy, J.C.G. Calado, W.B. Streett, Chem. Eng. Commun. 6, 365-383 (1980)
A. Hientz, W.B. Streett, Ber. Bunsenges. Phys. Chem. 87, 298-303 (1983)
W.B. Streett, J.C.G. Calado, J. Chem. Thermodyn. 10, 1089-1100 (1978)
W.B. Streett, Icarus 29, 173-186 (1976)
W.B. Streett, Astrophys. J. 186, 1107-1125 (1973)
M.L. McGlashan, K. Stead, C. Warr, J. Chem. Soc. Faraday Trans. 2, 1889–1895 (1977)
M.S. Green, M. Vicentini-Missoni, J.M.H.L. Sengers, Phys. Rev. Lett. 18, 1113-1117 (1967)
Acknowledgements
The authors acknowledge Dr Ziqing Pan and Luc Paoli for their contributions to the development of the internal PR EoS implementation.
Funding
No funds, grants, or other support were received for this study.
Author information
Authors and Affiliations
Contributions
RVL adapted the apparatus, carried out the measurements, processed the experimental data, performed model optimisation and comparisons, and drafted the initial manuscript. JPMT supervised the work and guided the overall investigation. Both authors contributed to the development of the PR EoS implementation, edited the manuscript, and reviewed the final version.
Corresponding author
Ethics declarations
Competing interest
The authors have no competing interests to declare.
Ethical Approval
Not applicable to this work.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Selected Papers of the 22nd European Conference on Thermophysical Properties.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Latcham, R.V., Trusler, J.P.M. Phase Behavior of Isobutane + CO2 and Isobutane + H2 at Temperatures Between 190 and 400 K and at Pressures Up to 20 MPa. Int J Thermophys 45, 13 (2024). https://doi.org/10.1007/s10765-023-03304-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10765-023-03304-0