
Abstract
To identify genes important for colorectal cancer (CRC) development and metastasis, we established a new metastatic mouse organoid model using Sleeping Beauty (SB) transposon mutagenesis. Intestinal organoids derived from mice carrying actively mobilizing SB transposons, an activating KrasG12D, and an inactivating ApcΔ716 allele, were transplanted to immunodeficient mice. While 66.7% of mice developed primary tumors, 7.6% also developed metastatic tumors. Analysis of SB insertion sites in tumors identified numerous candidate cancer genes (CCGs) identified previously in intestinal SB screens performed in vivo, in addition to new CCGs, such as Slit2 and Atxn1. Metastatic tumors from the same mouse were clonally related to each other and to primary tumors, as evidenced by the transposon insertion site. To provide functional validation, we knocked out Slit2, Atxn1, and Cdkn2a in mouse tumor organoids and transplanted to mice. Tumor development was promoted when these gene were knocked out, demonstrating that these are potent tumor suppressors. Cdkn2a knockout cells also metastasized to the liver in 100% of the mice, demonstrating that Cdkn2a loss confers metastatic ability. Our organoid model thus provides a new approach that can be used to understand the evolutionary forces driving CRC metastasis and a rich resource to uncover CCGs promoting CRC.
Similar content being viewed by others


Introduction
Colorectal cancer (CRC) is the third leading cause of cancer-related deaths worldwide, with metastatic CRC (mCRC) being the deadliest form of the disease. Recent multi-omics analyses of CRC have identified numerous genes important for CRC and helped us to understand the complexity of CRC progression [1, 2]. For example, targeted sequencing of metastatic CRC for genes that are included in the MSK-IMPACT panel, showed that mutations in TP53 are more frequent in mCRC than in primary tumors [3]. This study provided a dataset of genes mutated in primary CRC and mCRC, however, the genetic events driving mCRC are still largely unknown.
Sleeping Beauty (SB) transposon mutagenesis provides a genetic approach for the genome-wide identification of cancer genes in mice [4, 5]. Several SB mutagenesis screens have been performed in the intestine of wild-type mice, and in the intestine of mice carrying mutations in genes important for human CRC. This has led to the identification of hundreds of candidate CRC genes, providing a rich resource with which to study the function of genes involved in the development and progression of CRC [6,7,8,9,10,11]. Moreover, functional validation of several candidate tumor suppressors identified in these screens, such as Zfp292, Arid2, Acvr2a, Acvr1b, Trim33, and Arhgap5 has been performed [9, 12, 13], confirming the value of SB screens for identifying genes driving human CRC. Modeling mCRC using SB mutagenesis has, however, proven to be difficult due to the early death of mice caused by the increased tumor burden. Therefore, new experimental systems are needed to genetically dissect mCRC.
In this study, we established a new mouse model of mCRC using organoids derived from mice carrying actively mobilizing SB transposons, in addition to mutations in genes known to be important for CRC, such as Apc and Kras. Intestinal organoids are known to maintain physiological functions of intestinal epithelial cells in vitro [14]. Therefore, when transplanted to mice, these organoids can induce tumors that are histopathologically like tumors that develop naturally in vivo [15]. To immortalize intestinal organoids and enhance their tumor-forming ability, we also cultured them in the absence of growth factors known to be important for the maintenance of intestinal epithelial-derived organoids.
To identify candidate cancer genes (CCGs) insertionally mutated by SB in tumors, we established a new in-house informatics pipeline to detect common transposon insertion sites (CISs). CISs are regions in the genome of tumor cells that contain a statistically higher number of transposon insertions than would occur by random chance. Analysis of CISs and linked genes in primary tumors and metastatic tumors provided us with a list of CCGs responsible for tumor development. These datasets also give insights into the evolutionary processes driving tumor development and a rich resource of genes deregulated in mCRC. Finally, we validated three of these genes, Cdkn2a, Atxn1, and Slit2 in tumor development and showed that Cdkn2 is also a metastasis suppressor gene.
Results
Establishment of an organoid mouse model for metastatic CRC using SB mutagenesis
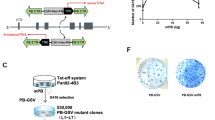
To identify genes involved in metastatic CRC, we mobilized SB transposons in mouse organoids carrying a loss-of-function mutation in Apc and an activating mutation in Kras. To produce these organoids, we first generated compound mutant mice carrying 5 different mutant alleles: lsl-SB11, T2/Onc2, Villin-CreERT2, ApcΔ716/+ [16] and lsl-KrasG12D/+ [17] (AK-SB mice, Fig. 1A). lsl-SB11 knock-in mice carry a cDNA for the SB transposase together with an upstream lox-STOP-lox transcriptional stop cassette inserted into the ubiquitously expressed mouse Rosa26 locus [6]. T2/Onc2 transgenic mice carry >300 copies of the SB transposon all linked together at a single site in the genome [18]. Villin-CreERT2 is an intestinal epithelial-specific inducible Cre line [19]. To facilitate the immortalization of mouse organoids and tumor development, we also incorporated a loss-of-function mutation in Apc (ApcΔ716) [16] and an activating point mutation in Kras (KrasG12D). We chose these mutant alleles because APC and KRAS mutations are observed at high frequency in human CRC (> 80% and >40% of human CRC, respectively). We then established organoids from both normal and tumor epithelial cells of AK-SB mice (Fig. 1B).

A Experimental design used to establish a xenograft mouse model for metastatic CRC using colonic organoids derived from AK-SB mice. B A photo for AK-SB organoids. C AK-SB organoids were transplanted to the cecum of anesthetized NSG mice using a needle (Black arrow). D, E Macroscopic appearance of tumors developed in the cecum. F A mouse developed both a cecum tumor (Black arrow) and metastatic liver tumors (White arrows). G, H Metastatic liver tumors were observed as shown by arrows. I Macroscopic appearance of peritoneal disseminated tumors (Black arrows). J A low magnification image of an HE stained primary tumor developing in the cecum. Enlarged pictures of regions in J by open squares are shown in K. L HE staining of a peritoneally disseminated tumor. M HE staining of a tumor metastasized to the liver. Bars; 0.5 mm for B and J, 5 mm for D, E, G and H, 1 cm for F and I, 100 μm for K, L and M.
To immortalize intestinal organoids, we cultured them in the absence of R-spondin, Noggin, and EGF, which are important for the maintenance of intestinal epithelial-derived organoids. Immortalized AK-SB organoids were then transplanted to the serosal side of the cecum [20] using a 30 G bent needle, taking care not to damage blood vessels (Fig. 1C). Three to six months later, mouse necropsies were performed. Forty-four out of 66 mice (66.7%) developed primary tumors in the mucosa of the cecum (Fig. 1D, E, and Table 1). Five mice (7.6%) developed both primary tumors and metastatic tumors in the liver, and also exhibited peritoneal disseminations (Fig. 1F–I). Histological analyses showed that most primary tumors were well or moderately differentiated adenocarcinomas (Fig. 1J, K). Peritoneally disseminated (PD) tumors (Fig. 1L) and liver tumors (Fig. 1M) were composed of gland like structures or sarcomatous colorectal cancers, thus modeling human CRC.
Identification of genes responsible for tumor development
To identify genes responsible for tumor development, we collected 38 primary tumors, 21 metastatic liver tumors and 31 peritoneal disseminations. We also collected 41 pre-injected organoid samples (Table 2). We then cloned the SB integration sites from these samples by ligation-mediated PCR and performed next generation sequencing, as previously described [9, 21]. Sequence reads were then mapped to the mouse genome, and candidate cancer genes (CCGs) were identified using our in-house informatics pipeline [22].
From this analysis, we identified 640 CCGs from primary tumors, 76 CCGs from metastatic liver tumors, 178 CCGs from peritoneal disseminations, and 91 genes from pre-injected organoids (Table 2, Fig. 2A, Supplementary Tables 1–6). The organoids were generated from 2 mice; normal colonic organoids were established from 1 mouse, and 14 tumor organoids were established from 1 mouse (Supplementary Fig. 1). To determine whether these tumors are independent of each other, we performed clustering analysis based on Hamming distance (version 1.7–4; http://cran.r-project.org/web/packages/e1071) [23] for organoids and primary tumors. Clustering analysis showed that the organoids used for transplantation were independent of each other, even if they were derived from the same mouse (Supplementary Fig. 2). This is consistent with a previous study, which showed that intestinal tumors developing in the same mouse were also independent of each other. We also performed the same clustering analysis for 38 primary tumors and found that these primary tumors were also independent of each other (Supplementary Fig. 3), even if the tumors were derived from the same organoids. Collectively, these data showed that organoids acquire new insertional mutations important for tumor development during growth in vivo.

A A Venn diagram showing the number of overlapping CCGs identified from primary tumors, metastatic liver tumors, peritoneal disseminations (PD) and organoids by SB screening. B A Venn diagram showing the comparison of CCGs identified in previous SB screens in the intestine (SB_CRC) and this study. C Pathway analysis using genes identified from primary tumors. D A Venn diagram showing the number of overlapping genes identified from Primary tumors in this SB screen and genes mutated in human CRC.
Next, we compared our list of CCGs identified from primary tumors with the dataset obtained from previous SB screens in the intestine [9, 22]. The number of overlapping genes was 404, and the number was statistically significant (p < 0.001. Fig. 2B, Supplementary Table 7), showing that our organoid transplantation model for CRC faithfully recapitulates SB screens performed in vivo.
Pathway analyses for CCGs identified in primary tumors showed that these genes function in many cancer-related pathways, including ‘Colorectal cancer’, ‘Hippo signaling’ and ‘TGF-β signaling’ (Fig. 2C and Supplementary Table 8). For CCGs identified from metastatic liver tumors, four pathways predominated, including ‘Pathways in cancer’ and ‘MicroRNAs (Supplementary Table 9). Likewise, CCGs identified in peritoneal disseminations were enriched in five pathways, including ‘Endocytosis’, ‘Ubiquitin mediated proteolysis’ and ‘Ras signaling’ (Supplementary Table 10). Collectively, pathway analyses showed CCGs identified from SB screens are enriched for genes that function in several cancer-causing signaling pathways.
The most frequently mutated gene identified in primary tumors was Apc (Supplementary Table 1), which is the primary gatekeeper in human and mouse intestinal cancer [6, 7, 9]. Presumably, SB insertions in primary tumors are in the wild type Apc allele present in organoid cells and result in the complete loss of Apc expression. In addition, Arhgap5 and Trp53 are also highly mutated in primary tumors, consistent with previous in vivo SB screens performed in the mouse intestine [9]. These data show that the orthotopic transplantation model using mouse organoids recapitulates human CRC as well as SB-induced intestinal cancer.
The second most frequently mutated gene identified in primary tumors was Cdkn2a, with 68.4% of primary tumors carrying mutations in Cdkn2a. CDKN2A functions as a tumor suppressor gene in various tissues, including the pancreas [24, 25], hematopoietic system [26] and skin [27]. CDKN2A encodes 2 different genes, p16 and p14 (p19 in mice) and functions in cell senescence.
Comparison of mouse CCGs with genes important for human CRC
Next, 624 non-redundant CCGs identified from primary tumors, metastatic liver tumors and peritoneal disseminations were compared to genes mutated in human CRC. Genes mutated in ≥ 3% of human CRC were selected and used for comparison. The number of overlapping genes was 211, and the number was statistically significant (p < 0.001, Fig. 2D, Supplementary Table 11). These genes included APC, TP53, FBXW7 and ACVR1B, which are well established CRC driver genes, showing that CCGs identified in our organoid transplantation model are significantly enriched for genes important for human CRC development.
Identification of CCGs involved in tumor metastasis
Since metastatic tumors were not genetically independent of each other (Supplementary Figs. 4 and 5), metastatic liver CCGs identified from only one mouse were removed to generate a final gene list (Supplementary Tables 2 and 3). Subsequently, we focused on 6 genes commonly mutated in primary tumors, peritoneal disseminations, and metastatic liver tumors, since mutations in these genes were considered likely to confer the ability to metastasize to remote organs and help the cells adapt to new environments. These genes are Ppm1l, Top2b, Atxn1, Cdkn2a, Hnrnpr and Atl2 (Fig. 3A, Supplementary Table 6). Protein phosphatase 1-like (Ppm1l) is a member of the serine/threonine phosphatase PP2C family. Ppm1l knockdown in macrophages promotes DAMP-triggered inflammatory cytokine production, showing that Ppm1l can be involved in inflammatory responses [28]. Top2b gene encodes a topoisomerase. A study showed that down-regulation of TOP2B was involved in enhancing chemosensitivity to oxaliplatin in CRC [29], in contrast, TOP2B mutations were observed in ovarian cancers [30] suggesting the cancer suppressive role of TOP2B. Atxn1 is a chromatin-binding factor that represses Notch signaling in the absence of the Notch intracellular domain by acting as a CBF1 corepressor [31]. Genetic alterations in Atxn1 have been observed in 20 of 447 (4%) cases of CRC [1]. Among these 20 cases, 7 appeared to represent deletions, frame-shifts, or nonsense mutations [32] and are likely to represent loss-of-function mutations. Furthermore, analysis of a dataset consisting of 148 microarray samples showed that ATXN1 mRNA expression is downregulated in stage I, II, III, and IV CRC [33]. These data suggest that the deregulation of ATXN1 is potentially involved in CRC development as well as metastasis. Hnrnpr encodes an RNA-binding protein that is a member of the spliceosome C complex. A study showed that Hnrnpr was downregulated in glioblastomas that acquired resistance to Temozolomide, the first-line chemotherapy agent [34], suggesting the involvement of Hnrnpr in relapse of glioblastoma. Atl2, which encodes Atlastin GTPase 2, was differentially expressed in breast cancers with bone metastases [35], raising the possibility that Atl2 was involved in metastasis. Finally, we also identified several other genes exclusively mutated in metastatic tumors (Supplementary Table 6) and these genes could also give survival advantages in remote organs and be involved in tumor metastases.

A An oncoplot for genes identified from Primary T, Liver met. and PD. B Genomic position of transposon insertions in Cdkn2a identified from organoids, Primary T, Liver met. and PD. Bars indicate transposon insertions. C Hamming distance between a primary tumor and metastatic tumors in mouse #320.
Primary tumors and metastatic tumors are clonally related
Among the 6 genes that are commonly mutated in primary tumors, peritoneal disseminations and metastatic liver tumors, we focused on Cdkn2a because of its high mutation frequency in each cohort (Fig. 3B). Interestingly, Cdkn2a was not mutated in organoids (Fig. 3B), suggesting that loss of Cdkn2a expression is not required for organoid survival. In sharp contrast, Cdkn2a was frequently mutated in primary tumors and metastatic tumors. Moreover, SB transposons were inserted at the exact same genomic position in metastatic tumors and primary tumors from the same mouse (Fig. 3B), showing that metastatic tumors were indeed derived from primary tumors. The SB transposon is known to mobilize relatively at random throughout the genome, except that they are inserted into TA sites [5]. The insertion of SB transposons is also less likely to be affected by chromatin status or the presence or absence of functional genetic loci on the genome [36]. These data indicate that loss of Cdkn2a is necessary for the development of primary tumors as well as metastatic tumors. Clustering analyses based on Hamming distance for mouse #320 showed that insertional profiles in metastatic tumors were more similar between metastatic tumors compared to primary tumors from the same mouse (Fig. 3C and Supplementary Fig. 6). These data show that metastatic tumors are clonally related to each other.
Functional validation of Cdkn2a, Atxn1 and Slit2 in tumor organoids
Although Cdkn2a is a well-established tumor suppressor gene, previous studies showed that homozygous Cdkn2a deletion in ApcMin mice had no effect on tumor number in the small intestine [37], suggesting that the tumor suppressive function of Cdkn2a in Apc-inactivated cells are very weak or nonexistent. In contrast, homozygous knockout of Cdkn2a in epithelial cells of BrafV600E/+, BrafV637E/+, or KrasG12D/+ mice resulted in the development of serrated adenocarcinomas in the colon [38,39,40]. These data suggest that Cdkn2a can function as a tumor suppressor in intestinal cells carrying an intact Apc gene in cases where Ras signaling is activated. To confirm and extend these results, we knocked out Cdkn2a in mouse AK organoids using CRISPR-Cas9. We then subcutaneously transplanted these organoids to NSG mice and monitored the effect on tumor development (Fig. 4A). These studies showed that tumor formation was accelerated in AK organoids carrying Cdkn2a gRNA compared to AK organoids carrying non-targeted (nonT) gRNA, showing that Cdkn2a can function as a tumor suppressor in Apc- and Kras-mutated colonic epithelial cells (Fig. 4B, C). Subsequently, we confirmed the gene editing efficiency of the gRNAs by sequencing the genomic loci targeted by these gRNAs (Supplementary Fig. 7, Supplementary Table 12) since no good antibodies for mouse Cdkn2a were available.

A An experimental design to knockout candidate tumor suppressor genes in AK-Cas9 organoids by CRISPR-Cas9, and validate the oncogenic ability by subcutaneous transplantation. Subcutaneous primary tumor development was promoted by disruption of Cdkn2a (B), Atxn1 (D), and Slit2 (F) in AK-Cas9 organoids. *P < 0.05 by two-sided t-test. Data are represented as mean values +/− SD. HE staining of tumors derived from AK-Cas9 organoids carrying gRNA for Cdkn2a (C), Atxn1 (E), and Slit2 (G). H GSEA in AK-Atxn1KO vs AK-nonT. Bars; 50 μm for C, E, and G.
Atxn1 was mutated in both primary and metastatic tumors in our SB organoid screen (Supplementary Table 2). Additionally, 3.4% of human CRC contains a mutation in ATXN1, suggesting that Atxn1 is also an important driver gene in mouse and human CRC. Slit2 was identified from liver metastases in our present SB screen. In humans, termination mutations in SLIT2 or deep deletion of the SLIT2 locus were observed in ~1% of CRC, implicating a tumor suppressor. To functionally validate these results, we knocked out Atxn1 or Slit2 in AK organoids using CRISPR-Cas9 and monitored the effect on tumor formation in NSG mice (Fig. 4A). The efficiency of tumor development in NSG mice transplanted with AK organoids carrying Atxn1 gRNAs or Slit2 gRNAs was significantly increased compared with mice transplanted with AK organoids carrying non-targeting control gRNA, which rarely caused tumor development (Fig. 4D–G). We confirmed the gene editing efficiency of the gRNAs by sequencing the genomic loci targeted by these gRNAs (Supplementary Figs. 8, 9 and Supplementary Table 12). These results show that Atxn1 can also function as a tumor suppressor gene in colonic epithelial cells of mice carrying mutations in Apc and Kras.
To understand the possible molecular mechanisms of Atxn1 in CRC development, we performed RNA-seq using AK-Atxn1-KO and AK-nonT control organoids. The GSEA showed that signals such as ‘G2M checkpoint’ and ‘E2F targets’ were activated in AK-Atxn1-KO organoids (Fig. 4E), suggesting the involvement of Atxn1 in cell cycle control.
Functional validation of metastasis-associated genes
Since Cdkn2a and Atxn1 were frequently mutated in metastatic tumors, we decided to determine whether the loss of these genes is important for tumor metastasis by transplanting CRISPR-Cas9 genetically engineered organoids to the spleen (Fig. 5A). We used spleen transplantation since this is one of the standard approaches used for rapid validation of the metastatic ability of tumor cells. When Cdkn2a KO organoids were transplanted to the spleen, tumors metastasized to the liver in only 6 weeks following transplantation, in 100% of the transplanted mice, while Atxn1 KO organoids never metastasized to the liver (Fig. 5B, C). These data show that loss of Cdkn2a function can confer the metastatic potential to AK organoids, while loss of function mutations in Atxn1 can not. Furthermore, we generated double knockout AK organoids carrying gRNAs for Cdkn2a and Atxn1 (Atxn1KO:Cdkn2aKO AK organoids) and transplanted them in the spleen. Mice transplanted with Atxn1KO:Cdkn2aKO AK organoids developed larger liver tumors compared with Cdkn2aKO organoids (Fig. 5D–H), showing that loss of Atxn1 conferred growth advantages to the cells as suggested by the GESA analysis (Fig. 4H), after colonization of the liver.

A An experimental design used to validate the metastatic-promoting ability resulting from Cdkn2a loss. B Comparison in the number of liver metastases between Atxn1KO, Cdkn2aKO, and Atxn1KO:Cdkn2aKO. *P < 0.05 by two-sided t-test. Data are represented as mean values +/− SD. C HE staining of metastatic liver tumors developed in the mouse transplanted with Atxn1KO organoids. D HE staining of metastatic liver tumors developed in the mouse transplanted with Atxn1KO:Cdkn2aKO organoids. E Macroscopic appearance of the liver of the mouse transplanted with Atxn1KO:Cdkn2aKO organoids. Arrows are metastatic foci. F HE staining of a metastatic liver focus developed in the mouse transplanted with Atxn1KO:Cdkn2aKO organoids. G Immunohistochemistry for E-cadherin in the liver focus developed in the mouse transplanted with Atxn1KO:Cdkn2aKO organoids. Bars: 200 μm for C and D, 5 mm for E, 100 μm for F, and 50 μm for G. H Incidence of mice with met. area ≥10% of liver area. Two-sided Fisher’s exact test.
Discussion
In this study, we generated an organoid model for metastatic CRC using SB mutagenesis and identified CCGs that could function in CRC development and metastasis. One of the most frequently mutated genes in primary tumors and metastatic tumors was Cdkn2a. Interestingly, Cdkn2a was not identified as a highly mutated gene in SB screens performed previously in the intestine [6, 7, 9, 10], but frequently mutated in our SB screens performed in colitis mice. The detailed analyses further showed that cancer cells in TNFα rich inflammatory microenvironment strongly select Cdkn2a mutations to gain growth advantage [22]. Likewise, in human CRC, CDKN2A is mutated or deleted in only 2.5% of CRC according to the TCGA dataset [1], while the promoter in the methylation of CDKN2A was a relatively common early event in colitis-associated cancers [41, 42].
One possibility for frequent mutations in Cdkn2a in our organoid model is that Cdkn2a inactivation increases the engraftment potential of organoids in the cecum of NSG mice, and organoids carrying insertions in Cdkn2a were preferentially selected. This is consistent with a previous study showing that a subpopulation of pediatric glioma cells carrying a CDKN2A deletion expanded when transplanted to a mouse intracranially [43], suggesting that Cdkn2a loss may be advantageous to the engraftment of glioma cells. These findings suggest that the high frequency of Cdkn2a mutations may be highly dependent on the cancer microenvironment.
We also demonstrated the metastatic ability of Cdkn2a loss in colon tumor cells, however, human cancer genome sequencing studies have infrequently identified mutations in CDKN2A in CRC. It is known, however, that the promoter of CDKN2A is hypermethylated in 30% of tumors, and that promoter hypermethylation is correlated with lymphovascular invasion and lymph node metastasis [44]. Thus, epigenetic silencing of CDKN2A in CRC [45] may be what’s responsible for promoting metastases. Consistent with this, recent whole-exome sequencing of brain metastases from lung adenocarcinomas identified a genomic region with significantly more frequent deletions in CDKN2A/B compared to nonmetastatic lung adenocarcinomas [46].
Organoids have been extensively used in cancer research to study genetic events involved in cancer development or drug resistance. These studies show that organoids more faithfully recapitulate the dynamics of cancer cells compared to ordinary two-dimensional cultures. By transplanting organoids to mice, metastatic processes can be modeled [47]. Taking advantage of this, we identified candidate genes associated with cancer metastasis, validated three genes in CRC, and showed that Cdkn2a loss confers the metastatic capacity to colonic cells.
In this study, we show that our organoid model provides a unique and powerful approach to identify novel metastatic CRC genes. By comparing these genes and those identified in human cancer genome sequencing, we may one day have a clearer picture of the genes driving mCRC and better therapeutics for treating this deadly disease.
Material and methods
Mice
NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ female mice were purchased from Charles River. Mice were anesthetized by Isoflurane (Wako) inhalation. Around a 1 cm incision was made on the skin of the lower abdomen, and the cecum was pulled out. Under a stereoscopic microscope, 3 ×105 cells dissolved in 30 μl of Matrigel (Corning) were injected into the subserosal layer of the cecum using a 30 G bent needle, trying not to hurt vessels [48]. The wounds were closed using an AutoClip applier (FST). All animal experimental protocols were approved by the committee on Animal Experimentation (Study number: AP-163819) of Kanazawa University or National Cancer Center Institute (Study number: T19-006-M07).
Organoid culture
Colonic organoids were established from AK-SB mice carrying lox-STOP-lox-SB11 [6], T2/Onc2 [18], Villin-CreERT2 [19], ApcΔ 716/+ [16] and KrasG12D/+ [17] as previously described [12]. Organoids were cultured in organoid medium which contained 50% of L-WRN conditioned medium [49], 20% fetal bovine serum (Biosera), penicillin/streptomycin, GlutaMax (Invitrogen), 10 nM of Y-27632 (Wako), 5 nM of A83-01 (Tocris) in advanced DMEM-F12 (Invitrogen). Tumor organoids were also established from intestinal tumors of AK-SB mice, and maintained in AK medium which contained 10 mM HEPES, GlutaMax, penicillin/streptomycin, N2 (Invitrogen), B27 (Invitrogen), 50 ng/ml EGF (Peprotech), 100 ng/ml Noggin (Peprotech) and 1 μM N-acetylcystein in advanced DMEM-F12 (Invitrogen). Pictures of organoids were obtained using a stereoscopic microscope (LEICA M205 FCA) with a LEICA DFC 7000T camera (Leica).
Histology
Tumor tissues were fixed in 4% paraformaldehyde overnight and embedded in paraffin blocks. Four μm sections were made and stained with Hematoxylin and Eosin. For immunohistochemistry, the standard protocol described in the previous paper [50] was used. Images were taken using BZ-X810 (Keyence) or IX73 operated by the CellSens software (Olympus). Using ‘Hybrid Cell Count’ mode of BZ-X800 Analyzer software, the number of liver foci, and the proportion of the area for liver foci in the liver on the slide were calculated.
SB library and NGS
Genomes from organoids were isolated using LaboPass Blood Mini (Hokkaido System Science). Tumor genomes were purified using a Gentra Puregene Tissue Kit (QIAGEN). For preparation of SB libraries, we followed the protocol previously described [9]. Genomes were digested with NlaIII or BfaI and ligated with NlaIII linkers or BfaI linkers. Linker ligated genomes were PCR amplified using IRDR(R1): GCTTGTGGAAGGCTACTCGAAATGTTTGACCC or IRDR(L1): CTGGAATTTTCCAAGCTGTTTAAAGGCACAGTCAAC, and Linker primer: GTAATACGACTCACTATAGGGC. Then second PCR was performed using IRDR_nested_illumina:TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGGTGTATGTAAACTTCCGACTTCAAC, Linker-nested llumina: GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGAGGGCTCCGCTTAAGGGAC. PCR products were labeled using a Nextera XT Index Kit v2 Set A and B (Illumina), and sequenced on a sequencer (Illumina). Detailed information for each sample in the SB library is described in Supplementary Tables 13 and 14.
Identification of CCGs
All sequencing reads were aligned to the mouse reference genome and candidate cancer genes were detected as described in our previous paper [22].
For pathway analysis, an online tool, DAVID [51] (https://david.ncifcrf.gov/) was used.
Validation of CCGs in NSG mice
To generate gRNA vectors, gRNAs targeting Cdkn2a (gRNA-1:GGTGGTGCTGCACGGGTCA, gRNA-2:CCGGGCGGGAGAAGGTAGT) or Atxn1 (gRNA-1:CAGCAGACCACGCATCGAG, gRNA-2:AGCAGTCGTGATACCTCCG) or Slit2 (gRNA-1: TTCGCCAAAGGCCACGGGT, gRNA-2:ATCCGCTAGCCACTTGAGA) were cloned into lentiviral vector, pKLV2.0 [52]. The non-target gRNA vector was described previously [12]. For generation of lentivirus, HEK293FT cells (ATCC Cat# PTA-5077, RRID:CVCL_6911) were plated on polylysine-coated dishes in high-glucose DMEM (Invitrogen). The pKLV2.0 carrying gRNA sequences were co-transfected with packaging vectors (ViraPower Lentiviral Expression Systems, Invitrogen) using polyethylenimine. On the next day, the culture medium was replaced with 5 ml of high-glucose DMEM. Ninety-six hours later from transfection, viral supernatants were collected and concentrated using Lenti-X (Takara). These viral particles were transduced into colonic AK organoids carrying Cas9 [12] which was generated previously. Transduction of lentivirus into organoids were described previously [12]. After 2 weeks of puromycin selection, 1 ×105 cells were transplanted subcutaneously or into the spleen of anesthetized mice. Knockout efficiency was confirmed by cloning the gRNA targeting genomic loci using PCR primers provided in Supplementary Table 15 and sequencing the PCR products with next-generation sequencing. The sequence results are presented in Supplementary Table 12, demonstrating the successful achievement of gene knockout (Supplementary Table 12 and Supplementary Figs. 7–11).
RNA sequencing
Organoid RNA was collected using ISOGEN (Nippon Gene) and treated with DNaseI. 1.5 μg of RNA was used for RNA-seq. The library was prepared using NEBNext UltraTMII Directional RNA Library Prep Kit (Cat No. E7760) following the manufacturer’s protocol, and sequenced on the illumine Novaseq 6000 by PE150. Reference sequences for mapping were prepared using rsem-prepare-reference of RSEM version 1.3.1 and STAR version 2.7.7a. Sequence reads were trimmed using trim_galore of Trim Galore version 0.6.7 and Cutadapt version 3.5. The processed reads were mapped to the reference sequences and TPM (Transcripts Per Million) was calculated using rsem-calculate-expression of RSEM version 1.3.1 and STAR version 2.7.10a. Gene and isoform TPM values were extracted from the TPM columns of RSEM result files. GRCm39 genome build was used for the RNA-seq analyses. The accession number for the data is GSE248668.
For GSEA analysis, the software, GSEA_4.3.2, was used. A Chip platform (Mouse_Gene_Symbol_Remapping_MSigDB.v2023.1.Mm.chip) was downloaded from the GSEA website.
Statistics and reproducibility
For Figs. 4B, D, F and 5B, data are represented as mean values +/− SD, statistical test was performed by two-sided t-test. Experiments were independently performed at least three times for Figs. 4B, D, F and 5B (Cdkn2aKO) and two times for Fig. 5B (Atxn1KO and Atxn1KO:Cdkn2aKO). No statistical method was used to predetermine sample size, but sample size was set to obtain the maximum results with the minimum number of samples to perform statistical analyses. No data were excluded from the analyses. The investigators were not blinded to allocation during experiments and outcome assessment for all experiments. For animal experiments, no randomization was used.
Data availability
The RNA-seq data in this study have been deposited in the Gene Expression Omnibus (GEO) database under accession codes GSE248668.
References
TCGA. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–7.
Giannakis M, Mu XJ, Shukla SA, Qian ZR, Cohen O, Nishihara R, et al. Genomic Correlates of Immune-Cell Infiltrates in Colorectal Carcinoma. Cell Rep. 2016;15:857–65.
Yaeger R, Chatila WK, Lipsyc MD, Hechtman JF, Cercek A, Sanchez-Vega F, et al. Clinical Sequencing Defines the Genomic Landscape of Metastatic Colorectal Cancer. Cancer Cell. 2018;33:125–136.e3.
Weber J, Braun CJ, Saur D, Rad R. In vivo functional screening for systems-level integrative cancer genomics. Nat Rev. Cancer. 2020;20:573–93.
Copeland NG, Jenkins NA. Harnessing transposons for cancer gene discovery. Nat Rev Cancer. 2010;10:696–706.
Starr TK, Allaei R, Silverstein KA, Staggs RA, Sarver AL, Bergemann TL, et al. A transposon-based genetic screen in mice identifies genes altered in colorectal cancer. Science. 2009;323:1747–50.
March HN, Rust AG, Wright NA, Ten Hoeve J, de Ridder J, Eldridge M, et al. Insertional mutagenesis identifies multiple networks of cooperating genes driving intestinal tumorigenesis. Nat Genet. 2011;43:1202–9.
Starr TK, Scott PM, Marsh BM, Zhao L, Than BL, O’Sullivan MG, et al. A Sleeping Beauty transposon-mediated screen identifies murine susceptibility genes for adenomatous polyposis coli (Apc)-dependent intestinal tumorigenesis. Proc Natl Acad Sci USA. 2011;108:5765–70.
Takeda H, Wei Z, Koso H, Rust AG, Yew CC, Mann MB, et al. Transposon mutagenesis identifies genes and evolutionary forces driving gastrointestinal tract tumor progression. Nat Genet. 2015;47:142–50.
Morris SM, Davison J, Carter KT, O’Leary RM, Trobridge P, Knoblaugh SE, et al. Transposon mutagenesis identifies candidate genes that cooperate with loss of transforming growth factor-beta signaling in mouse intestinal neoplasms. Int J Cancer. 2017;140:853–63.
Takeda H, Jenkins NA, Copeland NG. Identification of cancer driver genes using Sleeping Beauty transposon mutagenesis. Cancer Sci. 2021;112:2089–96.
Takeda H, Kataoka S, Nakayama M, Ali MAE, Oshima H, Yamamoto D, et al. CRISPR-Cas9-mediated gene knockout in intestinal tumor organoids provides functional validation for colorectal cancer driver genes. Proc Natl Acad Sci USA. 2019;116:15635–44.
Takeda H. A Platform for Validating Colorectal Cancer Driver Genes Using Mouse Organoids. Front Genet. 2021;12:698771.
Sato, Vries T, Snippert RG, van de Wetering HJ, Barker M, Stange DE N, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009;459:262–5.
Lee SH, Hu W, Matulay JT, Silva MV, Owczarek TB, Kim K, et al. Tumor Evolution and Drug Response in Patient-Derived Organoid Models of Bladder Cancer. Cell. 2018;173:515–28.e17.
Oshima M, Oshima H, Kitagawa K, Kobayashi M, Itakura C, Taketo M. Loss of Apc heterozygosity and abnormal tissue building in nascent intestinal polyps in mice carrying a truncated Apc gene. Proc Natl Acad Sci USA. 1995;92:4482–6.
Haigis KM, Kendall KR, Wang Y, Cheung A, Haigis MC, Glickman JN, et al. Differential effects of oncogenic K-Ras and N-Ras on proliferation, differentiation and tumor progression in the colon. Nat Genet. 2008;40:600–8.
Dupuy AJ, Akagi K, Largaespada DA, Copeland NG, Jenkins NA. Mammalian mutagenesis using a highly mobile somatic Sleeping Beauty transposon system. Nature. 2005;436:221–6.
el Marjou F, Janssen KP, Chang BH, Li M, Hindie V, Chan L, et al. Tissue-specific and inducible Cre-mediated recombination in the gut epithelium. genesis. 2004;39:186–93.
Fumagalli A, Suijkerbuijk SJE, Begthel H, Beerling E, Oost KC, Snippert HJ, et al. A surgical orthotopic organoid transplantation approach in mice to visualize and study colorectal cancer progression. Nat Protoc. 2018;13:235–47.
Mann KM, Ward JM, Yew CC, Kovochich A, Dawson DW, Black MA, et al. Sleeping Beauty mutagenesis reveals cooperating mutations and pathways in pancreatic adenocarcinoma. Proc Natl Acad Sci USA. 2012;109:5934–41.
Shimomura K, Hattori N, Iida N, Muranaka Y, Sato K, Shiraishi Y, et al. Sleeping Beauty transposon mutagenesis identified genes and pathways involved in inflammation-associated colon tumor development. Nat Commun. 2023;14:6514.
Meyer D, Leisch F, Hornik K. The support vector machine under test. Neurocomputing. 2003;55:169–86.
Habbe N, Langer P, Sina-Frey M, Bartsch DK. Familial pancreatic cancer syndromes. Endocrinol Metab Clin North Am. 2006;35:417–30.
Kamisawa T, Wood LD, Itoi T, Takaori K. Pancreatic cancer. Lancet. 2016;388:73–85.
Williams RT, Sherr CJ. The INK4-ARF (CDKN2A/B) locus in hematopoiesis and BCR-ABL-induced leukemias. Cold Spring Harb Symp Quant Biol. 2008;73:461–7.
Leachman SA, Carucci J, Kohlmann W, Banks KC, Asgari MM, Bergman W, et al. Selection criteria for genetic assessment of patients with familial melanoma. J Am Acad Dermatol. 2009;61:677.e1–14.
Wang B, Zhou Q, Bi Y, Zhou W, Zeng Q, Liu Z, et al. Phosphatase PPM1L Prevents Excessive Inflammatory Responses and Cardiac Dysfunction after Myocardial Infarction by Inhibiting IKKβ Activation. J Immunol. 2019;203:1338–47.
Zhang YJ, Li AJ, Han Y, Yin L, Lin MB. Inhibition of Girdin enhances chemosensitivity of colorectal cancer cells to oxaliplatin. World J Gastroenterol : WJG. 2014;20:8229–36.
Beltrame L, Di Marino M, Fruscio R, Calura E, Chapman B, Clivio L, et al. Profiling cancer gene mutations in longitudinal epithelial ovarian cancer biopsies by targeted next-generation sequencing: a retrospective study. Ann Oncol. 2015;26:1363–71.
Tong X, Gui H, Jin F, Heck BW, Lin P, Ma J, et al. Ataxin-1 and Brother of ataxin-1 are components of the Notch signalling pathway. EMBO Rep. 2011;12:428–35.
Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–4.
Asghari M, Abazari MF, Bokharaei H, Aleagha MN, Poortahmasebi V, Askari H, et al. Key genes and regulatory networks involved in the initiation, progression and invasion of colorectal cancer. Fut Sci OA. 2018;4:FSO278.
Yi G-Z, Xiang W, Feng W-Y, Chen Z-Y, Li Y-M, Deng S-Z, et al. Identification of Key Candidate Proteins and Pathways Associated with Temozolomide Resistance in Glioblastoma Based on Subcellular Proteomics and Bioinformatical Analysis. Biomed Res Int. 2018;2018:5238760–5238760.
Savci-Heijink CD, Halfwerk H, Koster J, van de Vijver MJ. A novel gene expression signature for bone metastasis in breast carcinomas. Breast Cancer Res Treat. 2016;156:249–59.
Yoshida J, Akagi K, Misawa R, Kokubu C, Takeda J, Horie K. Chromatin states shape insertion profiles of the piggyBac, Tol2 and Sleeping Beauty transposons and murine leukemia virus. Sci Rep. 2017;7:43613.
Cole AM, Ridgway RA, Derkits SE, Parry L, Barker N, Clevers H, et al. p21 loss blocks senescence following Apc loss and provokes tumourigenesis in the renal but not the intestinal epithelium. EMBO Mol Med. 2010;2:472–86.
Carragher LA, Snell KR, Giblett SM, Aldridge VS, Patel B, Cook SJ, et al. V600EBraf induces gastrointestinal crypt senescence and promotes tumour progression through enhanced CpG methylation of p16INK4a. EMBO Mol Med. 2010;2:458–71.
Rad R, Cadinanos J, Rad L, Varela I, Strong A, Kriegl L, et al. A genetic progression model of Braf(V600E)-induced intestinal tumorigenesis reveals targets for therapeutic intervention. Cancer Cell. 2013;24:15–29.
Bennecke M, Kriegl L, Bajbouj M, Retzlaff K, Robine S, Jung A, et al. Ink4a/Arf and oncogene-induced senescence prevent tumor progression during alternative colorectal tumorigenesis. Cancer Cell. 2010;18:135–46.
Sato F, Harpaz N, Shibata D, Xu Y, Yin J, Mori Y, et al. Hypermethylation of the p14(ARF) gene in ulcerative colitis-associated colorectal carcinogenesis. Cancer Res. 2002;62:1148–51.
Hsieh CJ, Klump B, Holzmann K, Borchard F, Gregor M, Porschen R. Hypermethylation of the p16INK4a promoter in colectomy specimens of patients with long-standing and extensive ulcerative colitis. Cancer Res. 1998;58:3942–5.
Kogiso M, Qi L, Lindsay H, Huang Y, Zhao X, Liu Z, et al. Xenotransplantation of pediatric low grade gliomas confirms the enrichment of BRAF V600E mutation and preservation of CDKN2A deletion in a novel orthotopic xenograft mouse model of progressive pleomorphic xanthoastrocytoma. Oncotarget. 2017;8:87455–71.
Xing X, Cai W, Shi H, Wang Y, Li M, Jiao J, et al. The prognostic value of CDKN2A hypermethylation in colorectal cancer: a meta-analysis. Br J cancer. 2013;108:2542–8.
Shima K, Nosho K, Baba Y, Cantor M, Meyerhardt JA, Giovannucci EL, et al. Prognostic significance of CDKN2A (p16) promoter methylation and loss of expression in 902 colorectal cancers: Cohort study and literature review. Int J Cancer. 2011;128:1080–94.
Shih DJH, Nayyar N, Bihun I, Dagogo-Jack I, Gill CM, Aquilanti E, et al. Genomic characterization of human brain metastases identifies drivers of metastatic lung adenocarcinoma. Nat Genet. 2020;52:371–7.
Fujii M, Shimokawa M, Date S, Takano A, Matano M, Nanki K, et al. A Colorectal Tumor Organoid Library Demonstrates Progressive Loss of Niche Factor Requirements during Tumorigenesis. Cell Stem Cell. 2016;18:827–38.
Céspedes MV, Espina C, García-Cabezas MA, Trias M, Boluda A, Gómez del Pulgar MT, et al. Orthotopic microinjection of human colon cancer cells in nude mice induces tumor foci in all clinically relevant metastatic sites. Am J Pathol. 2007;170:1077–85.
Miyoshi H, Ajima R, Luo CT, Yamaguchi TP, Stappenbeck TS. Wnt5a potentiates TGF-beta signaling to promote colonic crypt regeneration after tissue injury. Science. 2012;338:108–13.
Sakai E, Nakayama M, Oshima H, Kouyama Y, Niida A, Fujii S, et al. Combined mutation of Apc, Kras and Tgfbr2 effectively drives metastasis of intestinal cancer. Cancer Res. 2017;78:1334–46.
Da Huang W, Sherman BT, Stephens R, Baseler MW, Lane HC, Lempicki RA. DAVID gene ID conversion tool. Bioinformation. 2008;2:428–30.
Tzelepis K, Koike-Yusa H, De Braekeleer E, Li Y, Metzakopian E, Dovey OM, et al. A CRISPR Dropout Screen Identifies Genetic Vulnerabilities and Therapeutic Targets in Acute Myeloid Leukemia. Cell Rep. 2016;17:1193–205.
Acknowledgements
We thank the TCGA Research Network for sharing the CRC somatic mutational data. We also thank S. Kataoka, A. Tsuda, M. Watanabe, Y. Jomen and K. Sato for technical assistance. Histological sections were made by Y. Shiotani in the Core facility of National Cancer Center Research Institute. This study was supported by FOREST (JPMJFR2164), JSPS (21K19421, 20H03522), the National Cancer Center Research and Development Fund (2020-A-5), Princess Takamatsu Cancer Fund, Takeda Science Foundation, AMED (23ama221529h0001) and JH (2022-B-02).
Author information
Authors and Affiliations
Contributions
NI and YS established the informatics pipeline for SB screening and generated the SB data. YM and J-WP performed the animal experiments, analyzed the data. MO provided the mouse lines and established organoids. SS diagnosed the tumor. NAJ and NGC provided the mouse lines and wrote the manuscript. HT designed the experiment, analyzed the data, and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Iida, N., Muranaka, Y., Park, J.W. et al. Sleeping Beauty transposon mutagenesis in mouse intestinal organoids identifies genes involved in tumor progression and metastasis. Cancer Gene Ther 31, 527–536 (2024). https://doi.org/10.1038/s41417-023-00723-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41417-023-00723-x
