
Abstract
The combination of trimethoprim and sulfamethoxazole as a fixed-dose combination in the ratio 1:5 is known as cotrimoxazole. It is used as antibiotic to treat a variety of bacterial infections. Cotrimoxazole is part of the World Health Organization’s list of essential medicines. Cotrimoxazole is an example of a drug that was partially unavailable in Germany during the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) pandemic and the Ukraine war. The dependency on foreign sources of medicines is well known in Europe and resulted in the Pharmaceutical Strategy for Europe 2020, a strategy concept “will support the competitiveness and innovative capacity of the EU’s pharmaceutical industry”. High-performance thin-layer chromatography (HPTLC) is a cost-effective method for quantifying pharmaceutically active compounds. Diode-array detection (DAD) in conjunction with HPTLC can simultaneously detect ultraviolet‒visible (UV‒VIS) and fluorescence spectra directly from the plate. Visualization as a contour plot helps to identify the optimal wavelengths for compound quantification and reduce uncertainty in the determination. The quantification of trimethoprim and sulfamethoxazole is presented in a case study that highlights the key aspects for HPTLC quantification of pharmaceutical fixed-dose combinations with minimal uncertainty. HPTLC‒DAD allows quantification of trimethoprim and sulfamethoxazole with a required relative standard deviation of less than 2.5%.
Similar content being viewed by others

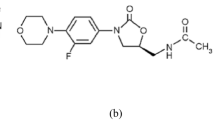

1 Introduction
The combination of trimethoprim and sulfamethoxazole as a fixed-dose combination in the ratio of 1:5 is known as cotrimoxazole. It is used as an antibiotic to treat a variety of bacterial infections. Cotrimoxazole is part of the World Health Organization’s list of essential medicines [1]. Trimethoprim is a 2,4-diaminopyrimidine derivative and blocks the dihydrofolate reductase in some bacteria. Sulfamethoxazole is a sulfonamide that prevents folic acid synthesis in some bacteria. Cotrimoxazole is an example of a typical drug that was not fully available in Germany during the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) pandemic. The dependency on foreign drug sources is well known in Europe and resulted in the Pharmaceutical Strategy for Europe 2020, a strategic concept that “will support the competitiveness and innovative capacity of the EU’s pharmaceutical industry” [2].
The analysis of trimethoprim and sulfamethoxazole in tablets or liquids is the subject of a large number of thin-layer chromatography (TLC) publications [3,4,5,6,7,8,9,10,11,12,13,14,15]. The tablet content must be within the range 95.0–105.0% of the specified amount [16]. A sophisticated assay limit analysis is necessary to verify this. Today, high-performance liquid chromatography (HPLC) dominates in Europe for limit assay analysis of drugs, although HPTLC has been shown to be less expensive than HPLC [17]. This paper is a case study for trimethoprim and sulfamethoxazole to show what is necessary to perform pharmaceutical limit analysis using modern HPTLC in combination with diode-array detection (DAD). This combination can simultaneously acquire ultraviolet‒visible (UV‒VIS) and fluorescence spectra directly from the plate. Visualization as a contour plot helps to determine the optimal wavelengths for compound quantification and reduces uncertainty in the determination. A two-dimensional evaluation of the plate is also possible.
The purpose of this work is to quantify trimethoprim and sulfamethoxazole in the 1:5 fixed-dose combination with minimal uncertainty and to find the minimum number of HPTLC tracks necessary to meet the objectives of ± 2.5% uncertainty in the final preparation.
2 Experimental
2.1 Preparation of sample and standards and application on the HPTLC plate
All the chemicals used were of analytical reagent grade. Trimethoprim was from Sigma Aldrich (Steinheim, Germany) and sulfamethoxazole was purchased from Sigma Chemicals (St. Louis, MO). Both standards have a purity of ≥ 99.5%. methanol, and cyclopentyl methyl ether (CPME) was from Carl Roth (Karlsruhe, Germany). Aluminum foil silica gel 60 F254 plates (1.05554) with a fluorescent dye, used as the stationary phase, were from Merck (Darmstadt, Germany). Cotrim-CT 800 mg/160 mg tablets were from CT Arzneimittel (Ulm, Germany, with batch number A54912) and were purchased from a local pharmacy.
Standard solutions were prepared by weighing the standards in different amounts (trimethoprim 1.608 mg and 2.855 mg) and sulfamethoxazole (11.529 mg and 11.850 mg) using an Orion Cahn® C-33 microbalance from Environmental Instruments (Beverly, MA). The standards were dissolved in 10 mL of methanol. The standard solutions of lower content were mixed at a 1:1 ratio to standard St1, and the higher standard solutions were mixed at a 1:1 ratio to standard St2. The final concentrations in St1 were 80.4 µg/mL for trimethoprim and 576.45 µg/mL for sulfamethoxazole. The final concentrations in St2 were 142.75 µg/mL for trimethoprim and 592.5 µg/mL for sulfamethoxazole.
Five cotrimoxazole tablets were weighed (1205.7 mg, 1197.5 mg, 1190.3 mg, 1194.0 mg, and 1194.7 mg) and mortared. An amount of 43.751 mg powdered cotrimoxazole was dissolved in 25 mL of methanol and sonicated for 10 min. The solution was abbreviated as S. The solution was centrifuged before plate application.
Standard and sample solutions were spotted band-wise over 7 mm on prewashed plates (prewashed with methanol–water with a ratio of 8:2, V/V) using the automatic TLC Sampler (ATS 4) from CAMAG (Muttenz, Switzerland) equipped with a 25 µL syringe. The bands were spotted at a distance of 10 mm from the bottom plate edge and at a distance of 0.5 cm from the plate edges. The sample solution S was applied in amounts of 3.5 µL, standard St1 in amounts of 5 µL, and St2 in amounts of 9 µL using the pair application technique.
2.2 Separation conditions
Silica gel plates (10 cm × 10 cm) were developed in the dark at 23 °C and 35% relative humidity in a vertical developing chamber (twin-trough glass chamber lined with filter paper) at vapor saturation (60 min) to a distance of 60 mm, calculated from the point of application with the solvent CPME–methanol–water (7.6:2:0.4, V/V). The plates were vapor-conditioned for 10 min prior to development using metal hair clips to suspend the plates in the vapor. Development was initiated by opening the clips so that the plates would drop 5 mm to stand in the solvent. The two plates (aluminum foil) were developed simultaneously over 6 cm in the same chamber, standing “surface to surface”.
2.3 Spectral measurements from plate
A TIDAS TLC S700 system from J&M (Aalen, Germany), with a reflection attachment consisting of three rows of optical fibres, was used for the spectral measurements of the plate, which has a wavelength resolution of 0.8 nm and a spatial resolution on the plate of 100 µm (produced by TransMIT-Centre for Fibre Optics and Industrial Laser Applications, Gießen, Germany). The middle row was used for detection, and the other two rows were connected to a deuterium lamp (for UV measurements) and a 365-nm light-emitting diode (LED) for fluorescence measurements. The measurement time for a single spectrum in the wavelength range from 190 to 1000 nm was 25 ms. For two-dimensional measurements, a reflection attachment was used with nine identical optical fibers with a diameter of 100 µm each (produced by TransMIT-Centre for Fibre Optics and Industrial Laser Applications). These fibers were used for illumination purposes and were arranged around a single fiber with a diameter of 200 µm. The spatial resolution on the plate was 200 µm. For fluorescence illumination, an LED (model: LEDMOD 365.1, produced by Omicron Laserage, Rodgau, Germany) was used instead of a mercury lamp. The wavelength-dependent reflectance (R) was calculated after measuring J and J0 according to the following equation:
where J is the intensity of light reflected and/or scattered from a sample track and J0 is the intensity of light reflected and/or scattered from a blank track.
The raw data of the measurement were evaluated using expression (2), which is the function of the extended Kubelka–Munk equation [18, 19].
where k is the backscattering factor (0 ≤ k ≤ 1), a is the absorption coefficient, J0 is the reflected light intensity measured from a neat plate part, and J is the reflected light intensity measured from a sample track.
The factors k and l adjust Eq. (2) to special measurement conditions. For example, in trace analysis, not too much light is absorbed by the analyte, and almost all of the illuminated light is reflected by from the plate surface. This is taken into account by setting the backscattering factor k in Eq. (2) to 1, leading to Eq. (4) [18, 19].
In the case of fluorescence, there is no need to consider backscattering, so Eq. (2) for k = 0 gives the desired transformation Eq. (5) [18,19,20].
3 Statistical requirements
The Annex to Council Directive of 26 October 1983, amending directives 65/65/EEC, 75/318/EEC, and 75/319/EEC, states: “Unless there is appropriate justification, the maximum acceptable deviation in the active substance content of the finished products shall not exceed ± 5% at the time of manufacture” [16]. In other words, the assay limit must be in the confidence interval between 95.0% and 105.0%. The confidence interval with a significance level of α = 0.05 can be interpreted as containing the true value with a probability of 95%.
Common practice worldwide is that half of the accepted uncertainty in the drug content is calculated for manufacturing and half for analysis. Thus, the task is to quantify trimethoprim and sulfamethoxazole with a deviation of less than ± 2.5% of the labeled amount, leading to the analytical objective of quantifying the real content of the tablet formulation within a confidence interval of {‒2.5% of the labeled content} ≤ labeled content ≤ {+ 2.5% of the labeled content}. To do this, it must be ensured that the measured HPTLC peak areas {y1, …, yn} are an independent, normally distributed set of data (i.e., have the shape of a Gauss distribution). This data set has the unknown parameters true mean μ and true variance σ2, which are unknown, because one cannot measure an infinite number of data. The true mean and true variance can be estimated by the mean (\(\overline{Y }\)) and variance (S2), which must be calculated from a set of independently processed samples, applied individually on the plate, and measured track by track after separation. It should be noted that both values belong to a Student’s t-distribution with f = n − 1 degrees of freedom and do not belong to a Gauss distribution!
To calculate the overall uncertainty of an analysis, its variance (S2) and mean (\(\overline{Y }\)) are calculated from the measured peak areas (yi) according to expressions (6) and (7):
The relative standard deviation (%RSD) is calculated from the variance and the mean as follows:
In the second step, the content of trimethoprim and sulfamethoxazole (e.g., in mg/tablet) must be calculated from the peak area data. For this purpose, a reference measurement—a calibration—must be performed. The variance of a linear calibration as reference method (Sr2) corresponds to Eq. (7), where \(\overline{Y }\) in this equation is defined as \(\overline{Y }\)=ax + b. The relative standard deviation of the content (Sc/\(\overline{X }\)) is the sum of the relative variances of n analyte measurements in the sample and m reference measurement, as shown in (9):
The area of the bell curve is 100% when the integration limits are set from −∞ to + ∞. To calculate a 95% confidence interval (cnf95%, with α = 0.05) for the real mean μ, one would have to integrate the Gauss function in ± 2σ limits (correct: ± 1.96 σ)! In this case, there is a 2.5% probability that the value is smaller and a 2.5% probability that it is larger than the 95% confidence interval. As mentioned above, neither the value of µ nor of σ is known, but we can estimate it from S2 and \(\overline{Y }\) using Student’s t-distribution. The Student factor tα,f depends on the degree of freedom f and the level of significance α and is the factor between the Gaussian and Student’s distribution, which describes Eq. (10):
The confidence interval of the final result with the limits \(\overline{X }-{\text{cnf}}_{95\%}\) and \(\overline{X }+{\text{cnf}}_{95\%}\) can be interpreted as containing the real mean µ with a probability of 95%. Assuming that the relative variances of sample (\({S}_{s}^{2}/{\overline{Y} }_{s}^{2}\)) and reference (\({S}_{r}^{2}/{\overline{Y} }_{r}^{2}\)) are identical to (\({S}^{2}/{\overline{Y} }^{2}\)) and f = n − 1 = m − 2 (which means that we use a linear regression equation with two parameters as calibration function); the confidence interval can be calculated from Eq. (10) using Eqs. (8) and (9) as follows:
Figure 1 shows the values of the square root in Eq. (11) as (cnf95%/%RSD), depending on the degrees of freedom. When using a linear regression as calibration, expression (11) is valid only for sample values which lie exactly in the middle of the calibration graph.

Reduced confidence intervals for different degrees of freedom, calculated without %RSD for f = n − 1 sample and f = m − 2 standard measurements with equal variance (S) of standard and sample according to Eq. (11). The final uncertainty in the confidence interval is calculated from the reduced confidence interval multiplied by the achievable relative standard deviation (%RSD). To achieve the accepted uncertainty of 2.5%, a %RSD of at least 1.17% is required for f = 3, while a %RSD of 2.18% is sufficient for f = 7
A 10 cm × 10 cm HPTLC plate provides space for nine tracks, where four sample and five calibration tracks should be used. To perform the analysis for f = 3 (n = 4 and m = 5) with an overall deviation of < 2.5%,, a relative standard deviation of S < 1.17% is required (Fig. 1). A relative standard deviation of S < 2.18 is required for f = 7 (n = 8 and m = 9) to obtain the maximum acceptable deviation.
4 Chromatographic requirements
It is not trivial to generate a chromatographic method with such low uncertainty, but some publication may help [21,22,23,24]. In particular, the ICH guidelines issued by the International Council for Harmonisation of Technical Requirements for Pharmaceuticals [21] are often cited as a quality standard, so that the term “according to ICH guidelines” seems to guarantee the required quality. In this way, ICH guidelines are completely misunderstood, because “the ICH guideline does not provide specific instructions how to validate different analytical methods” [24]. The introduction to guideline Q2(R1) clearly describes its aim: “The objective of validation of an analytical procedure is to demonstrate that the analytical procedure is suitable for the intended purpose.” The purpose of the present work is to demonstrate that the analytical method is suitable for the intended purpose of quantifying trimethoprim and sulfamethoxazole in a pharmaceutical preparation with an acceptable deviation of less than 2.5% and how this can be accomplished.
4.1 Chromatographic deviations
In chromatography, the analyte is distributed between the mobile and the stationary phase, resulting in an analyte distribution of unknown shape. For quantitative HPTLC, it is not necessary to know the distribution of the analyte in the layer. A constant analyte distribution for all sample and standard zones is essential for quantification. To achieve this, the stationary and mobile phase conditions must be constant for all tracks. As the vapor phase also influences the composition of the mobile phase, which is a result of the solvent distribution in the stationary and vapor phase, the conditions in all three phases must be constant for all analytes with the same RF values during the entire development process [25].
4.1.1 Solvent optimization
Modern HPTLC should avoid the use of hazardous solvents. However, this is not the case, as shown by the European Pharmacopoeia 10th Edition, where sulfamethoxazole is separated with the mobile phase: dilute ammonia–water–nitromethane–dioxane (3:5:41:51, V/V) [26]. A literature survey reveals that from 13 papers on the analysis of trimethoprim and sulfamethoxazole, 11 papers use CHCl3 as part of the mobile phase [5,6,7,8,9,10,11,12,13,14,15]. Only two publications avoid CHCl3 and instead use a mixture of toluene–ethyl acetate–methanol [4] or a mixture of ethyl acetate–methanol (3:1, V/V), in which trimethoprim unfortunately tends to tail strongly [3].
In the past, we have been successful in replacing CHCl3 by CPME [27], and indeed, a solvent mixture of CPME–methanol (9:2.9, V/V) can separate trimethoprim from sulfamethoxazole (RF = 0.76), with an RF value of 0.16. The disadvantage of this solvent is that trimethoprim shows a large tailing, which makes integration problematic. Sulfamethoxazole moves to a position near the front and peak integration is difficult if the plate is not clean, so a prewash step is mandatory.
The solvent CPME–methanol (9:2.9, V/V) shows that constant phase conditions are present for the trimethoprim peak, which stays close to the application zone. It can be quantified with low uncertainty without chamber saturation being accounted. The sulfamethoxazole zone moves near the front and cannot be quantified with a low uncertainty without chamber saturation. Obviously, the conditions near the solvent surface are constant even in a chamber without a presaturated vapor phase. For the sulfamethoxazole zone at a high RF value, this is not the case, so presaturation of the vapor phase by the solvent is required, using a chamber whose sides are covered with filter paper, which soaks up the solvent and provides a larger surface area for evaporation. It is also helpful to reduce the overall separation distance, and the RF value of the sulfamethoxazole zone can be reduced by preconditioning the plate in the chamber vapor for 10 min.
The solvent mixture CPME–methanol–formamide (8:1.5:0.5, V/V), with the RF values 0.2 and 0.77, avoids tailing of trimethoprim and keeps the trimethoprim band at a larger distance from the application point. Unfortunately, formamide remains entirely in the stationary phase, causing an uneven baseline and making a proper peak integration difficult. The best result was obtained by replacing formamide with water.
In a chamber with a presaturated vapor phase, the solvent mix CPME–methanol–water (7.6:2:0.4, V/V) shows a good separation, no tailing of the peaks, a low RF value of sulfamethoxazole, and a uniform baseline. The densitogram and the structure of both compounds are given in Fig. 2. Both peaks have a sufficiently large distance to the point of application (RF value of trimethoprim is 0.17) and the front signal (RF value of sulfamethoxazole is 0.55). It is important that the plate is prewashed with the solvent methanol–water (8:2, V/V) prior to separation to ensure a uniform baseline.

Densitogram of trimethoprim (left) and sulfamethoxazole (right) on silica gel 60 aluminum foil, separated with the solvent CPME–methanol–water (7.6:2:0.4, V/V)
4.1.2 Sample application
When selecting the track width of a separation, it must be taken into account that the light fiber width of the diode-array scanner is 3.5 mm. In other words, the sample application must have a homogenous analyte distribution of at least 3.5 mm in length on all tracks. This requires a band-wise application of at least 7 mm. Scanning in the middle of the band allows a constant measurement with uniform sample distribution, even with slight deviations from a perpendicular development. A spot-wise application with slight deviation from a perpendicular development would result in larger scanning deviations. In addition, a band-wise application in sharp bands allows a large application volume without loss of resolution. Keeping a distance of 3 mm to the next band gives track dimensions of 10 mm width.
A CAMAG ATS 4 device was used for band-wise sample application. CAMAG specifies the syringe resolution with 16 bits, thus the program can apply the entire syringe volume in 216 increments. Accepting an application uncertainty of 0.1% for 1 µL (which means a minimum of 1000 steps volume resolution) and using a 500 µL syringe, a rough estimation according to equation (12) shows that this only works if the application volume is larger than 7.6 µL:
Application of at least 1.53 µL would provide the required application uncertainty with a 100 µL syringe, as would an application of more than 0.38 µL with a 25 µL syringe.
4.1.3 Stationary phase optimization
The stationary phase must have a constant thickness across all tracks. Glass plates (in the size of 10 cm × 20 cm or 10 cm × 10 cm) have a smaller thickness at the plate sides, which is known as the “side effect”. This results in more band broadening at plate sides and, thus, lower analyte concentration in the middle of the band compared with tracks with constant thickness. Therefore, a 10 cm × 10 cm glass plate allows separation of only eight tracks, while on a 10 cm × 10 cm aluminum foil, separation of up to nine tracks is possible.
4.1.4 Plate drying
After separation, the plate must be dried uniformly. During drying of the plate, the analyte in the stationary phase moves towards the plate surface. This increases the analyte concentration at plate surface while the mobile phase evaporates. This is what makes the drying process so important, because uneven drying of the plate will result in different sample distributions on different tracks. It is best to place the plate in a fume hood for 30 min without moving the air above the plate surface. One should never use a hair dryer to dry the plate!
4.2 Peak-purity check
The ICH guidelines state that “specificity/selectivity can be shown by demonstrating that the identification and/or quantitation of an analyte is not impacted by the presence of other substances (e.g., impurities, degradation products, related substances, matrix, or other components present in the operating environment)” [21]. The method presented is selective for trimethoprim and sulfamethoxazole when both compounds are completely separated, and no interfering matrix is present in the analyte zones. To confirm this, a peak-purity check for both compounds is necessary. This is best done using a diode-array scanner. Figure 3 shows the contour plot of a track on which 3.5 µL of a cotrimoxazole sample is separated. The scan was performed in a wavelength range from 200 to 400 nm and shows two symmetrical peaks without wavelength-dependent deformation. The symmetrical structure of both peaks indicates that the separation of trimethoprim, as well as sulfamethoxazole, is not affected by insufficient separation or by the matrix.

Contour plot of a track, separated with the solvent CPME–methanol–water (7.6:2:0.4, V/V) and measured in absorption according to Eq. (4). Trimethoprim and sulfamethoxazole show symmetric peaks
4.3 Fluorescence detection of trimethoprim
Oxidation of trimethoprim either by dipping in nitric acid [28] or by contact of the plate with oxygen in combination with 24 h daylight irradiation [29] results in a bright fluorescence, which can be used for quantification. Figure 4A shows the contour plot of a cotrimoxazole sample track measured in fluorescence using am LED for illumination that emits light at 365 nm. The fluorescence spectrum of trimethoprim is plotted at left and was evaluated in the wavelength range from 428 to 452 nm by bundling 34 diodes. The resulting peaks of trimethoprim were evaluated, and they showed a standard deviation of always more than 5%. The trimethoprim fluorescence is not suitable for quantification with the required uncertainty.

Contour plot of a track, separated with the solvent CPME–methanol–water (7.6:2:0.4, V/V) and measured in fluorescence (A), according to Eq. (5), and in remission (B), according to Eq. (1). The fluorescence spectrum of trimethoprim in 4A is shown on the left and the densitogram at 440 nm is shown at the top
5 Detection requirements
5.1 Wavelength selection
The most important sources of noise in a DAD measurement are the light source, the detector, the signal amplifier, and the A/D converter. The reflectance values R(λ) are calculated from two different spectra: the sample spectrum [J(λ)] and the spectrum of the pure plate [J0(λ)] according to Eq. (1). Both spectra contribute to the noise in R(λ). The reflectance of, e.g., R = 0.1, measured at a given wavelength, describes a situation in which only 10% of the incident light is not absorbed by the analyte but reflected and, thus, measured by the detector, resulting in a small value and thus a high measurement uncertainty in J. This is the case for zones with high sample concentrations. A reflectance of R = 0.9 describes a situation in which the analyte absorbs only 10% of the incident light and is therefore nearly invisible to the detector. The difference to J0 is very small. This also results in a high uncertainty when calculating R. It can be seen that both situations cause a high error in R, according to Eq. (1). Acceptable errors in R(λ) occur at reflectance values greater than 0.3 and less than 0.7. A reflectance of R = 0.5 means that half of the incident light is absorbed and half is reflected back to the detector. This scenario exhibits the lowest error in R and should be used for quantification [30].
In Fig. 4B, a contour plot of a sample separation is visualized with Rmax = 0.7. For the trimethoprim peak, a signal is visible only in the 280–291 nm range. Only here R values smaller than 0.7 can be measured. In the final method, plate measurements were performed in the absorption range from 284 to 288 nm by bundling the signals of 13 diodes.
5.2 Peak sampling frequency
The number of peak data points measured should be as large as possible to obtain a good approximation of the true peak area. In most cases, the peak area is simply calculated as the sum of the discrete data points in the peak limits and not reconstructed from the measured data points. For this procedure, theoretical predictions show that the minimum number of data points sampled over a peak to accurately (< 0.1%) measure a peak area is at least 25 [31].
The diode-array detector has a spatial resolution on the plate of better than 100 µm [32]. The required number of data points, especially for the small peak of trimethoprim, can be easily achieved. According to Fig. 5, the number of data points for the trimethoprim peak is more than 30. The number of data points for the sulfamethoxazole peak is more than 50.

Data points of the densitogram of trimethoprim (left) and sulfamethoxazole (right) separated with the solvent CPME–methanol–water (7.6:2:0.4, V/V). For trimethoprim at least 31 data points can be distinguished, and for sulfamethoxazole at least 50 data points can be distinguished
6 Calibration
Prior to quantification, the type of calibration and the working range of the method must be determined. A linear calibration (Y = ax + b with the two coefficients a and b) is more favorable than a polynomial calibration because a polynomial calibration is determined by at least three coefficients. The degree of freedom for m calibration measurements with linear fitting is f = m − 2,, while a polynomial calibration results in a degree of freedom with f ≤ m − 3.
Quantification with light is generally a highly nonlinear method. Light measurements in scattering media is ruled by the extended Kubelka–Munk equation. In trace analysis, a backscattering factor k = 1 is usually used. At higher analyte concentrations, the backscattering factor must be reduced due to the fact that a substantial part of light is absorbed by the analyte. Calibration measurements show that a backscattering factor of k = 0.7 provides a sufficiently wide range of linearity for both substances (Fig. 6). Trimethoprim can be quantified in a linear range of 4‒2600 ng and sulfamethoxazole in the range of 18‒7100 ng.

Calibration of trimethoprim (A) and sulfamethoxazole (B), evaluated with k = 1 (blue) and k = 0.7 (red). The linear range for trimethoprim is 4‒2600 ng (Y = 0.004173x + 0.0542, R2 = 0.9991) and for sulfamethoxazole 18‒7100 ng (Y = 0.005851x + 0.2169, R2 = 0.9994)
Allowing only R values for trimethoprim in the (partially critical) range of 0.8–0.6 and for sulfamethoxazole in the R range of 0.4–0.28, the linear range for trimethoprim is reduced to 400–1300 ng and for sulfamethoxazole to 2800–5400 ng. It is interesting to note that an unfavorable R value of 0.24 is measured for sulfamethoxazole at the peak maximum, thus sulfamethoxazole should not be quantified at its peak maximum.
A test on linearity is superfluous, because quantification using a linear calibration model with nonlinear data will never achieve an assay uncertainty of ± 2.5%. In general, the calibration model is a chosen tool for quantification. In addition to a linear calibration, one should always use a nonlinear calibration model to check whether it is superior to a linear model. For example, the nonlinearized data in Fig. 6 (blue lines) can be perfectly approximated by a first-order polynomial function, but this model is usually not superior to a linear model because three regression parameters are required. This reduces the degree of freedom unfavorably compared with the linear calibration model.
7 Precision
7.1 Sample precision
Concerning the term “precision,” the ICH guidelines state: “The standard deviation, relative standard deviation (coefficient of variation) and confidence interval should be reported for each type of precision investigated and be compatible with the specification limits” [21].
To measure precision, five tablets purchased from a local pharmacy were mortared, weighed (43.751 mg), and topped up to a volume of 25 mL. The sample solution S was applied in amounts of 3.5 µL, standard St1 in amounts of 5 µL and St2 in amounts of 9 µL using the pair application technique (Table 1).
The two plates (aluminum foils) were developed simultaneously over 5 cm in the same chamber, standing “surface to surface”. Table 1 shows the scanning results, which were evaluated according to Eq. (2) with the backscattering factor of k = 0.7, calculated according to [33]. The scanning results are visualized in Fig. 7.

Quantification in the reduced linear calibration range of trimethoprim (A) and sulfamethoxazole (B) using Eq. (2) with a backscattering factor of k = 0.7
Plate B shows that it is possible to keep the uncertainty below 2.5% by using four sample and five calibration tracks on a 10 cm × 10 cm plate. Plate A shows that this is not always the case. Only the combination of all measurements made on two plates guarantees the required maximum acceptable deviation, as Table 2 shows.
7.2 Homoskedasticity
The variances for a set of measurements must be constant over the entire working range (homoskedasticity) [24]. This can be easily checked by calculating the variances of St1 and St2 for both compounds at the beginning and end of the calibration range, as shown in Fig. 7. If equation var(St1) = var(St2) is valid, the quotient (TF) will be 1.
The test value TF is to be compared with the values of the F-distribution at a significance level of α = 0.05. With six (St2) and four (St1) measurements at the outer ends of the calibration range, the test value of 9.01 should not be exceeded, otherwise the calibration range must be reduced. Both compounds fulfill the test, as Eqs. (13) and (14) show.
8 Accuracy
The ICH guidelines define the term accuracy as follows: “The accuracy of an analytical procedure expresses the closeness of agreement between the value which is accepted either as a conventional true value or as an accepted reference value and the value measured.” “Accuracy should be established across the reportable range of an analytical procedure and is typically demonstrated through comparison of the measured results with an expected value. Accuracy should be demonstrated under regular test conditions of the analytical procedure (e.g., in the presence of sample matrix and using described sample preparation steps)” [21].
8.1 Standard addition
The usual way of determination accuracy (since there is usually no second, fully validated methods available that allows parallel testing) is the standard addition method for calculating recovery. It is important to note that recovery should characterize the entire analytical procedure, beginning with sample preparation and ending with calculation of the result [24].
Sample preparation is the most critical step affecting accuracy, because a loss of analyte can occur during the procedure. The method presented does not require any critical sample preparation because the tablets are simply mortared and dissolved in methanol and this solution is applied directly on plate. Performing a standard addition with the sample would therefore only prove once again that the method is linear.
8.2 Analyte stability during chromatography
A second source of error that affects accuracy arises from the chromatography itself. It must be checked that no degradation of the analyte occurs during the separation process. This is best done by two developments with the same solvent, where the second development is performed perpendicular to the first development. All compounds showing no degradation lie on the diagonal of the chromatogram (Fig. 8), while decompositions show spots outside the diagonal.

Two-dimensional DAD scan of trimethoprim and sulfamethoxazole developed in both directions with the solvent mixture CPME–methanol–water (7.6:2:0.4, V/V) without chamber saturation and evaluated at 286 nm (A) and 210 nm (B)
A sample amount of 3.5 µL was spot applied and separated in the dark. After drying, the plate was separated once again perpendicular to the first separation but now under daylight. The plate was scanned track-wise using the DAD scanner with a spatial resolution of 200 µL [34].
The two-dimensional DAD densitogram at 286 nm in Fig. 8a shows no degradation products, neither on the diagonal of the chromatogram nor next to it. A closer look at 210 nm shows that sulfamethoxazole is degraded, especially during the second development because additional signals outside the diagonal can be seen here. The extent of the degradation is rather small, but to avoid interfering quantification, the separation should be performed in the dark.
8.3 Analyte content in the final product
The analytical results were calculated in comparison with the theoretically expected value, as proposed by ICH guidelines: “The analytical procedure is applied to an analyte of known purity (e.g., a reference material, a well characterized impurity or a related substance) and the measured versus theoretically expected result is evaluated” [21].
One tablet with an average weight of 1196.44 mg contains the amount of 800 mg sulfamethoxazole and 160 mg trimethoprim. Calculated according to Eqs. (15) and (16), a sample application of 3.5 µL contains the amounts of 4095.58 ng sulfamethoxazole and 819.12 ng trimethoprim:
Compared with the result of the trimethoprim analysis (840.16 ng*100%/819.12 ng = 102.57%) and the sulfamethoxazole analysis (4211.48 ng*100%/4095.58 ng = 102.83%), the method can verify that the tablets contain both trimethoprim and sulfamethoxazole within the specified content deviation of ± 5%.
9 Conclusions
DAD‒HPTLC can simultaneously measure UV‒VIS and fluorescence spectra directly on plate, which are visualized in contour plots to support peak-purity testing of separated zones. Essential is the possibility to select an appropriate wavelength range for quantification, depending on remission values, which should lie between 0.3 and 0.7. DAD‒HPTLC can also scan two-dimensional separations to verify analyte stability during the development process, and DAD‒HPTLC can quantify analytes with less than 2.5% deviation. For example, plate B shows that it is possible to keep the uncertainty below 2.5% using four sample and five calibration tracks on a 10 cm × 10 cm plate. Plate A shows that this is not always the case.
From a statistical point of view, at least nine tracks (four sample and five standards) are necessary to achieve less than 2.5% uncertainty. For 100% probability of less than 2.5%, you need more than nine tracks! It can therefore be concluded that the DAD‒HPTLC method is capable to measure trimethoprim and sulfamethoxazole with a deviation of less than 2.5% in commercially available tablets, making the method suitable for limit value analyses in the pharmaceutical industry.
Abbreviations
- HPTLC:
-
High-performance thin-layer chromatography
- TLC:
-
Thin-layer chromatography
- DAD:
-
Diode-array detection
- CPME:
-
Cyclopentyl methyl ether
- R 2 :
-
Correlation coefficient
- α :
-
Level of significance
- f :
-
Degree of freedom
- %RSD:
-
Relative standard deviation (in %)
References
World Health Organization (2019) World Health Organization model list of essential medicines: 22st list 2021. https://www.who.int/publications/i/item/WHO-MHP-HPS-EML-2021.02
Pharmaceutical Strategy for Europe (2020) https://health.ec.europa.eu/system/files/2021-02/pharma-strategy_report_en_0.pdf
Hauk C, Boss M, Gabel J, Schäfermann S, Lensch HPA, Heide L (2022) An open-source smartphone app for the quantitative evaluation of thin-layer chromatographic analyses in medicine quality screening. Sci Rep 12:13433. https://doi.org/10.1038/s41598-022-17527-y
Shewiyo DH, Kaale E, Risha PG, Dejaegher B, Smeyers-Verbeke J, Heyden YV (2009) Development and validation of a normal-phase high-performance thin layer chromatographic method for the analysis of sulfamethoxazole and trimethoprim in co-trimoxazole tablets. J Chromatogr A 1216:7102–7107
Fang B, Lu FL, Jiang Y, Cong PJ, Zhang Y (1998) Simultaneous identification of sulphamethoxazole, sulphadiazine and trimethoprim in Zengxiao Lianhuan tablets. Yaowu Fenxi Zazhi 18:269
Yun K, Li G (1997) Determination of sulfamethoxazole and trimethoprim in serum and urine by thin-layer chromatography. Chin J Hosp Pharm (Zhongguo Yaoxue Zazhi) 17:123–124
Lalla JK, Bhat SU, Sandu NR, Shah MU (1997) HPTLC determination of sulphamethoxazole and trimethoprim (cotrimoxazole). Indian Drugs 34:275–282
Agbaba D, Radovic A, Vladimirov S, Zivanow-Stakic D (1996) Simultaneous TLC determination of cotrimoxazole and impurities of sulfanilamide and sulfanilic acid in pharmaceuticals. J Chromatogr Sci 34:460–464
Feng Y, Zhang SH, Zhang Y, Shen S (1994) Determination of three components of trimethoprim-sulfadiazine-sulfamethoxazole tablets by thin-layer chromatography. Chin J Chromatogr (Sepu) 12:364–366
Li Z, Shi T, Wu J (1992) Determination of sulfamethoxazole in compound injection by thin-layer chromatography. Chin J Pharm Ind (Zhongguo Yiyao Gongye Zazhi) 23:321–322
Datta K, Das SK (1988) Thin-layer chromatographic method for rapid quantification and identification of trimethoprim and sulphamethoxazole in pharmaceutical dosage forms. J Liq Chromatogr 11:3079–3089
Tomankova H, Vasatova M, Zyka J (1988) TLC-densitometric determination of sulphonamide chemotherapeuticals. Anal Lett 21:2227–2240
Li T (1988) Determination of sulphamethoxazole and trimethoprim in tablets by thin-layer chromatography. Chin J Pharm Anal 8:185–186
Pawelczyk E, Plotkowiak Z, Nogowska M (1987) Chromatographic-spectrophotometric determination of sulphamethoxazole and trimethoprim in Biseptol (POLFA) [co-trimoxazole] suspension. Farm Pol 43:9–12
Tammilehto SA (1985) High-performance thin-layer chromatographic determination of trimethoprim and sulfamethoxazole in pharmaceutical dosage forms. J Chromatogr 323:456–461
Council Directive of 26 October 1983c, amending Directives 65/65/EEC, 75/318/EEC and 75/319/EEC on the approximation of provisions laid down by law, regulation or administrative action relating to proprietary medicinal products (83/570/EEC). https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=celex%3A31983L0570
Renger B (1999) Benchmarking HPLC and HPTLC in pharmaceutical analysis. J Planar Chromatogr 12:58–60
Spangenberg B (2006) Does the Kubelka-Munk theory describe TLC evaluations correctly? J Planar Chromatogr 19:332–341
Spangenberg B, Poole CF, Weins C (2010) Quantitative thin-layer chromatography. A practical survey. Springer, Berlin
Spangenberg B, Weyandt-Spangenberg M (2004) Quantitative thin-layer chromatography using absorption and fluorescence spectroscopy. J Planar Chromatogr 17:164–168
International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use. ICH Harmonised Guideline: Validation of Analytical Procedures Q2(R2). Draft version, Endorsed on 24 March 2022 ICH, Q2(R1), https://database.ich.org/sites/default/files/ICH_Q2-R2_Document_Step2_Guideline_2022_0324.pdf
Ferenczi-Fodor K, Vegh Z, Nagy-Turak A, Renger B, Zeller M (2001) Validation and quality assurance of planar chromatographic procedures in pharmaceutical analysis. J AOAC Int 84:1265–1276
Renger B, Végh Z, Ferenczi-Fodor K (2011) Validation of thin layer and high performance thin layer chromatographic methods. J Chromatogr A 1218:2712–2721. https://doi.org/10.1016/j.chroma.2011.01.059
Ferenczi-Fodor K, Renger B, Végh Z (2010) The frustrated reviewer—recurrent failures in manuscripts describing validation of quantitative TLC/HPTLC procedures for analysis of pharmaceuticals. J Planar Chromatogr 23:173–179. https://doi.org/10.1556/JPC.23.2010.3.1
Geis F, Schlitt H, Klose A (1965) Zur Reproduzierbarkeit in der DC. Z Anal Chem 213:331–346
The European Pharmacopoeia, 10th Edition (2019) Council of Europe, 67075 Strasbourg Cedex
Spangenberg B (2022) New solvent systems to separate some estrogenically active compounds by high-performance thin-layer chromatography (HPTLC). J Planar Chromatogr 35:89–96
Schlöbe R, Thijssen HHW (1982) Quantitative thin-layer chromatography of trimethoprim and tetroxoprim using fluorescence densitometry. J Chromatogr 230:212–215
Sigel CW, Grace ME (1973) A new fluorescence assay of trimethoprin and metabolites using quantitative thin-layer chromatography. J Chromatogr A 80:111–116. https://doi.org/10.1016/S0021-9673(01)85355-3
Spangenberg B, Klein KF, Mannhardt J (2002) Proposals for error-reduction in planar chromatography. J Planar Chromatogr 15:207–212
Rossi DT (1988) A simplified method for evaluating sampling error in chromatographic data acquisition. J Chromatogr Sci 26:101–105. https://doi.org/10.1093/chromsci/26.3.101
Spangenberg B, Klein KF (2000) Fibre optical scanning with high resolution in thin-layer chromatography. J Chromatogr A 898:265–269
Anders B, Doll S, Spangenberg B (2021) A validated quantification of triclosan in toothpaste using high-performance thin-layer chromatography and a 48-bit fatbed scanner. J Planar Chromatogr 34:203–209. https://doi.org/10.1007/s00764-021-00108-6
Milz B, Klein KF, Spangenberg B (2012) Quantitative two-dimensional thin-layer chromatography using a diode-array detector. J Planar Chromatogr 25:493–497
Acknowledgements
The authors express their appreciation to Merck Company (Darmstadt, Germany) for kind support.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author declares that he has no conflict of interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Spangenberg, B. Quality control in pharmaceutical analysis by diode-array thin-layer chromatography: quantification of trimethoprim and sulfamethoxazole as a case study. JPC-J Planar Chromat 36, 377–391 (2023). https://doi.org/10.1007/s00764-023-00276-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00764-023-00276-7