
Abstract
Recently, kagome lattice materials have emerged as a new model material platform for discovering and engineering novel quantum phases of matter. In this work, we elucidate the driving mechanism of the \(\sqrt{{{3}}}\)×\(\sqrt{{{3}}}\) charge order in a newly discovered kagome metal ScV6Sn6. Through multimodal investigations combining angle-resolved photoemission spectroscopy, phonon dispersion calculations, and phase diagram study, we identify the central role of unstable planar Sn and Sc phonon modes, while the electronic instability and van Hove singularities originating from the V kagome lattice have a marginal influence. Our results highlight that the \(\sqrt{{{3}}}\)×\(\sqrt{{{3}}}\) charge order in ScV6Sn6 is fundamentally distinguished from the electronically driven 2 × 2 charge order in the canonical kagome system AV3Sb5, uncovering a new mechanism to induce symmetry-breaking phase transition in kagome lattice materials.
Similar content being viewed by others


Introduction
Strongly correlated systems and topological materials are two different research areas in condensed matter physics, while exotic electronic phenomena often appear at their intersection1. Kagome lattice, a two-dimensional network of corner-sharing triangles (Fig. 1a), naturally lies at this intersection thanks to the unique symmetry-protected electronic structure composed of Dirac fermions at K, van Hove singularities (vHS) at M, and a flat band across the whole Brillouin zone (Fig. 1a). On one hand, the diverging density of states at the van Hove singularities and flat band fillings can promote various correlated many-body ground states2,3,4,5,6. On the other hand, the linear band crossing at K and the quadratic band touching degeneracy at Γ can be a singular source of Berry curvature and nontrivial topology7,8,9. Accordingly, the kagome lattice materials offer a promising opportunity to discover novel electronic phenomena at the confluence of correlation and topology, and have attracted significant research interests during the past few years10,11,12,13,14,15,16,17.

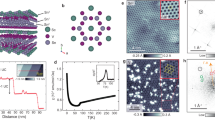
a Prototypical electronic structure of the kagome lattice featuring the Dirac point, vHS, and flat band. Inset displays the lattice structure. b, c Crystal structure of kagome metals CsV3Sb5 (a) and ScV6Sn6 (b) sharing the same V kagome plane. d, e Temperature-dependent X-ray diffraction profile and integrated peak area of the (2/3, 2/3, 20/3) charge order peak in ScV6Sn6, respectively. f Temperature-dependence of the in-plane resistivity of ScV6Sn6 around the charge order transition. g–j Various types of charge orders predicted from the extended Hubbard model on kagome lattice. k, l Real space mapping of the ARPES and Sn 4d XPS intensity of the ScV6Sn6 sample. Three different surface domains (D1, D2, and D3) with dramatically different ARPES and XPS spectra were identified. The representative D1, D2, and D3 domain positions are marked in (k, l. m–o), ARPES (Top panel) and XPS (bottom panel) of ScV6Sn6 on D1, D2, and D3 domains, respectively. ARPES spectra was obtained along K-Γ-K directions with 120 eV photons. The dashed cyan and orange boxes in (m–o) represent the area where the ARPES and XPS intensities are integrated and plotted in (k, l). The regions of integrations are chosen to maximize the contrast between different domains in (k, l).
A family of AV3Sb5 (A = K, Rb, Cs, Fig. 1b) represents an archetype kagome system hosting a rich series of emergent electronic orders, including the 2 × 2 charge order17, 1 × 4 stripe order18, electronic nematicity19, superconductivity17, and pair density waves20. The electronic instabilities associated with the vHSs of the V kagome lattice have been proposed as the origin behind these rich behaviors of AV3Sb521,22,23. Among these, the 2 × 2 charge order (Fig. 1g) exhibits many unconventional characteristics and has been considered a key to understand the exotic physics of AV3Sb5. For example, the 2 × 2 charge order in AV3Sb5 may accompany an imaginary conjugate component called chiral flux order24,25, which may explain the spontaneous time-reversal symmetry breaking26,27 and anomalous Hall conductivity28 observed in AV3Sb5 without explicit magnetism. Also, an intricate competition between the 2 × 2 charge order and superconductivity gives rise to the multiple superconducting domes in the phase diagrams of CsV3Sb529,30,31. In this context, understanding the nature of unconventional charge orders in kagome lattice materials is of fundamental importance in this emerging research field.
Meanwhile, the 2 × 2 charge order is not the only form of the charge order proposed in the kagome lattice: early theories predicted more diverse forms of charge order to appear at 1/3, 2/3, and vHS fillings, including not only the 2 × 2 charge bond order (Fig. 1g) but also the 1 × 1, 2 × 1, and \(\sqrt{3}\)×\(\sqrt{3}\) charge density waves (Fig. 1h–j)32,33,34,35. The latter phases are distinguished from the 2 × 2 charge bond order in AV3Sb5 by the charge disproportionation at each lattice site, which reflects the manifestation of long-range Coulomb interaction. Intriguingly, such charge disproportionation phases in the frustrated kagome lattice geometry have been predicted to host a fractional charge excitation e/233, analogous to the fractional spin excitations in kagome quantum spin liquids36. In this respect, exploring a new kagome system hosting diverse forms of charge order is highly desired, yet has been missing so far.
To this end, we turn our attention to the newly discovered kagome compound ScV6Sn6 hosting the novel \(\sqrt{3}\)×\(\sqrt{3}\) charge order below TCO ≈ 92 K (Fig. 1j)37. The ScV6Sn6 belongs to the large family of HfFe6Ge6-type ‘166’ kagome metals (Fig. 1c) with a prospect to tune the charge order by broad chemical substitutions16. However, the origin and nature of the \(\sqrt{3}\)×\(\sqrt{3}\) charge order in ScV6Sn6 have remained to be understood. On one side, the \(\sqrt{3}\)×\(\sqrt{3}\) charge order may be a consequence of the intrinsic electronic instability of the kagome lattice as predicted from the extended Hubbard model since the early 2010s (Fig. 1j)32,33,34. The ScV6Sn6 shares the partially filled V kagome lattice with the AV3Sb5, so it is tempting to suggest that the same vHS instability of AV3Sb5 also contributes to the charge order in ScV6Sn6. On the other side, the X-ray refinement of the charge order structure revealed the dominant displacement of the Sc and Sn atoms, while the displacement within the V kagome lattice is marginal37. Moreover, the \(\sqrt{3}\)×\(\sqrt{3}\) charge order is not generally observed in RV6Sn6 series (R = Sc, Y, and rare earth elements), suggesting that extrinsic factors specific to ScV6Sn6 may play a role.
In this work, we established the origin of the \(\sqrt{3}\)×\(\sqrt{3}\) charge order in ScV6Sn6 by comprehensively mapping its electronic structure, phonon dispersion, and phase diagram. Our multimodal approaches coherently point toward that the \(\sqrt{3}\)×\(\sqrt{3}\) charge order in ScV6Sn6 is tied to the structural components other than the V kagome lattice and is thus fundamentally different from the 2 × 2 charge order in AV3Sb5 originating from the intrinsic electronic instability of V kagome plane.
Results and discussion
Basic characterizations of ScV6Sn6
We start with the basic characterizations of ScV6Sn6. Our transport measurements revealed a sudden change in electrical resistivity around TCO ≈ 92 K, signaling a symmetry-breaking phase transition (Fig. 1f). X-ray diffraction measurements detected commensurate superlattice peaks in the low-temperature phase consistent with the \(\sqrt{3}\)×\(\sqrt{3}\) charge ordering (Fig. 1d). Both the abrupt drop in the diffraction peak intensity at TCO (Fig. 1e) and the small thermal hysteresis in resistivity (Fig. 1f) are indicative of the first-order nature of the transition. Overall, our transport and diffraction characterizations of ScV6Sn6 are in close agreement with the original report37.
Before discussing the detailed electronic structure of ScV6Sn6, we briefly remark on the possible surface terminations of the 166 kagome materials. As shown in Fig. 1c, the unit cell of ScV6Sn6 consists of one ScSn2 layer, one hexagonal Sn2 layer, and two V3Sn kagome layers; this HfFe6Ge6-type 166 structure can expose complex surface terminations upon cleaving. We note that previous studies on the 166 kagome materials yield inconsistent interpretations on the surface terminations (Supplementary Note 1). To resolve this issue, we performed spatially resolved ARPES and XPS experiments on ScV6Sn6 using micro-focused synchrotron radiation (Fig. 1k, l). As summarized in Fig. 1m–o, we clearly identified three different surface domains characterized by dramatically different valence band structures and Sn 4d3/2, 4d5/2 core level spectra (D1, D2, and D3 domains, respectively). By comparing the ARPES and XPS spectra at each domain to the slab DFT calculations of various geometries, we unambiguously assigned the D1, D2, and D3 domains to the ScSn2, V3Sn, and Sn2 surface terminations, respectively (Supplementary Note 2, 3). Below we focus on the results obtained on the V3Sn termination (D2), which best represents the bulk electronic structure of ScV6Sn6 based on the slab calculation (Supplementary Note 3).
van Hove singularities in ScV6Sn6
Figure 2 displays our analysis of the low-energy electronic structure of ScV6Sn6. Similar to the case of AV3Sb5, we identified multiple kagome-derived vHSs near the Fermi level. In Fig. 2a–c, we present a three-dimensional stack of the ARPES spectra measured at the vicinity of M point (see the momentum positions of the cut 1–4 in Fig. 2j, see also Supplementary Fig. 4 for the ARPES spectra in the extended momentum range). From these plots, one can comprehensively understand the dispersions along both the Γ-M-Γ (see solid lines in the cut 4) and the K-M-K direction (see dashed lines across the cut 1 to 4). As shown in Fig. 2a, b, we identified two bands having electron-like character along the K-M-K direction and hole-like character along the Γ-M-Γ direction; these bands thus form saddle point structures or vHSs at the M point as predicted from the model kagome lattice dispersion (see also schematics in Fig. 2g, h). As shown in Fig. 2c, we also observed one additional vHS with inverted concavity, i.e., hole-like dispersion along the K-M-K and electron-like dispersion along the Γ-M-Γ direction (see schematics in Fig. 2i). The density functional theory (DFT) calculations in Fig. 2d–f closely reproduce the experimental results, revealing that the three vHSs in Fig. 2a–c respectively originate from the dxy, dxz, and dz2 local orbital degrees of freedom in the V kagome lattice (Supplementary Note 4). In the kagome lattice, the sublattice character of vHS – pure (p) or mixed (m) sublattice character – is also a topic of great interest, which critically determines the relevance of the on-site and long-range Coulomb interactions and the leading electronic instabilities3,4,23. By analyzing the sublattice weight distribution near the M point, we revealed that all three vHSs in ScV6Sn6 are p-type vHs having pure sublattice character (Supplementary Note 5). In sum, our analysis provides the complete characterizations of the dispersions, orbital characters, and sublattice types of the vHSs in ScV6Sn6.

a, c Experimentally identified vHS dispersions in ScV6Sn6 using ARPES at 6 K. The cut 1–4 plot the ARPES spectra measured perpendicular to the K-M-K direction, with the cut 1 crossing the K point and the cut 4 crossing the M point (see panel j for the exact momentum positions of cut 1–4). We identified three coexisting vHS in ScV6Sn6 as marked with black, orange, and cyan guidelines in (a–c), respectively. d, e Corresponding DFT band structures of ScV6Sn6 for comparison with the ARPES spectra in a-c. The fat bands in d-f represent the spectral weight of the dxy, dxz, and dz2 local orbitals, respectively. g–i Schematics of the saddle point dispersions or vHSs. The concavity of vHS is identical for dxy and dxz vHS, while it becomes opposite for the dz2 vHS. j Schematics of the pristine (gray hexagons) and the \(\sqrt{3}\)×\(\sqrt{3}\) folded (cyan hexagons) in-plane Brillouin zones of the ScV6Sn6. Dashed and solid orange arrows represent the reciprocal lattice vectors. k–n Temperature dependence of the vHSs across the charge order transition. The cyan and black dashed lines are guide for the eye for the dz2 and dxy vHS dispersions near the Fermi level, respectively. All data were collected with 129 eV photons, measuring the kz ≈ 0 high-symmetry plane of the three-dimensional Brillouin zone.
Notably, the dxy and dz2 vHS of ScV6Sn6 locate very close to the Fermi level at –0.02 ± 0.01 eV and –0.03 ± 0.02 (Fig. 2a, c), while the dxz vHS is positioned at higher binding energy ≈ –0.40 ± 0.03 eV (Fig. 2b). The former vHSs contribute to the diverging density of states at the Fermi level and can in principle promote various electronic instabilities including the charge orderings. This scenario indeed applies to the case of AV3Sb5, where the vHSs at the Fermi level develop charge order gaps and directly contribute to the stabilization of the 2 × 2 charge order21,22,38,39. To test this scenario in ScV6Sn6, we tracked the temperature evolution of the vHSs across the \(\sqrt{3}\)×\(\sqrt{3}\) charge order transition. Comparing the ARPES dispersions in the normal (Fig. 2k, m) and charge-ordered states (Fig. 2l, n), we observed that all vHSs in ScV6Sn6 stay surprisingly unaltered across TCO, despite the fact that the vHS momentum (i.e., M point) lies at the folded Brillouin zone boundary of the \(\sqrt{3}\)×\(\sqrt{3}\) charge order (see the schematics in Fig. 2j). This observation indicates that in stark contrast to the case of AV3Sb5, the intrinsic electronic instability of the V kagome lattice plays a marginal role in driving the \(\sqrt{3}\)×\(\sqrt{3}\) charge order in ScV6Sn6.
Electronic reconstruction and charge order gap
After ruling out the vHS, we explore the electronic structure of ScV6Sn6 in the extended momentum range (Fig. 3), to identify the bands actually relevant to the \(\sqrt{3}\)×\(\sqrt{3}\) charge order transition. Figure 3a, b displays the Fermi surfaces of ScV6Sn6 measured at the normal and charge-ordered state, respectively. The major reconstruction of the Fermi surface across TCO is apparent from our data: the circular intensity pattern centered at \(\bar{\Gamma }\) in the normal state is modified to the star-shaped pattern in the charge-ordered state as highlighted with the cyan and orange guidelines. To better understand this change, we also present the corresponding energy-momentum dispersions along the \(\bar{\Gamma }\)-\(\bar{{\rm{M}}}\) direction in Fig. 3e, f. In the normal state dispersion (Fig. 3e), we observe a large electron pocket centered at \(\bar{\Gamma }\), which constructs the circular intensity pattern in the normal state Fermi surface (Fig. 3a). Below TCO (Fig. 3f), this electron band bends toward the higher binding energy and develops a substantial charge order gap at the Fermi level. This opening of the charge order gap depletes the intensity in the Fermi surface along the \(\bar{\Gamma }\)-\(\bar{{\rm{M}}}\) direction and explains the star-shaped Fermi surface observed in the charge-ordered state (Fig. 3b). The momentum position of the charge order gap is at about two-thirds of the \(\bar{\Gamma }\)-\(\bar{{\rm{M}}}\) direction, which excellently matches with the folded Brillouin zone boundary of the \(\sqrt{3}\)×\(\sqrt{3}\) phase (Fig. 2j). We note that the band renormalization and charge order gap is also observed in other surface terminations supporting their bulk origin (see Supplementary Fig. 8 for the charge order gaps measured in the D1 termination). As shown in Fig. 3g, h, our DFT calculations closely capture the experimental results, reproducing the large electron pocket at \(\bar{\Gamma }\) in the normal state (Fig. 3g) and opening of the charge order gap ΔCO ≈ 260 meV across the Fermi level in the charge order state (Fig. 3h). Notably, the magnitude of the charge order gap in ScV6Sn6 is significantly larger than the ΔCO ≈ 80 meV of AV3Sb5 despite the comparable TCO in two systems21,22.

a, b The Fermi surface of ScV6Sn6 in the normal and charge-ordered state, respectively. The data were obtained using 115 eV photons corresponding to the kz ≈ π high-symmetry plane. The cyan and orange solid lines in a-f are guide for the eye highlighting the band dispersion around the \(\bar{\Gamma }\) point. c, d Stack of the constant energy contours of ScV6Sn6 in the normal and charge-ordered state, respectively. e, f Normal and charge-ordered state dispersion of ScV6Sn6 measured along the \(\bar{\Gamma }\)-\(\bar{{\rm{M}}}\) high symmetry direction marked in (a, b). The cyan and orange arrows highlight the back bending of the dispersion due to the charge order gap opening at the Fermi level. The right panels in (e, f) display the energy distribution curves measured at the \(\sqrt{3}\)×\(\sqrt{3}\) charge order Brillouin zone boundary, i.e., at the two-thirds of \(\bar{\Gamma }\)-\(\bar{{\rm{M}}}\) momentum. The spectral weight shift and charge order gap opening is evident from the energy distribution curves as marked with the black arrow in (f). g, h DFT band structure of ScV6Sn6 in the normal and charge-ordered state, respectively. The band structure in the charge-ordered state is unfolded to the pristine Brillouin zone to facilitate comparison. The black arrow in (h) indicates the charge order gap observed in the experiment.
Microscopic origin of charge order in ScV6Sn6
Importantly, this large electron pocket at \(\bar{\Gamma }\), which is closely tied to the \(\sqrt{3}\)×\(\sqrt{3}\) charge order, has dominant planar Sn character (i.e., Sn(1) in Fig. 1c). To illustrate this, we present the DFT band structure of ScV6Sn6 in Fig. 4a, along with the V and Sn(1) orbital-projected calculations in Fig. 4b, c. The corresponding Fermi surfaces are also shown in the insets. In the V orbital-projected calculation (Fig. 4b), multiple Dirac bands at \(\bar{{\rm{K}}}\) and van Hove singularities at \(\bar{{\rm{M}}}\) originating from the V kagome lattice can be clearly identified. Overall, the V spectral weights dominate the Fermi surface near the zone boundary. In contrast, the Fermi surface near the zone center \(\bar{\Gamma }\) has dominant Sn(1) orbital character, as shown in the inset of Fig. 4c. We emphasize that it is this Sn(1) band at \(\bar{\Gamma }\) that develops the charge order gap and reconstructs the Fermi surface across TCO (Fig. 3), while the V kagome bands near \(\bar{{\rm{K}}}\) and \(\bar{{\rm{M}}}\) remain unaltered across TCO (Fig. 2). Our results thus highlight that the \(\sqrt{3}\)×\(\sqrt{3}\) charge order of ScV6Sn6 is tied to the structural components other than the V kagome lattice, especially to the planar Sn atoms.

a–c kz-integrated DFT band structure and its projection to the V and Sn(1) orbitals, respectively. The insets display the corresponding Fermi surfaces. d The phonon dispersion of ScV6Sn6 obtained from the DFPT calculation. e The phonon partial density of states of ScV6Sn6 projected to the Sc, V, Sn(1), Sn(2), and Sn(3) sites of the unit cell (Fig. 1c). f, g The DFPT phonon dispersion and phonon partial density of states of CsV3Sb5, respectively, for comparison with the ScV6Sn6 in (d, e).
The above conclusion from the electronic sector is further supported by our phonon mode calculations presented in Fig. 4d–g. As shown in Fig. 4d, the phonon dispersions of ScV6Sn6 display the continuum of unstable phonon modes centered at H, consistent with the \(\sqrt{3}\)×\(\sqrt{3}\) reconstruction in ScV6Sn6 at low temperature. By projecting the phonon density of states to the Sc, V, and Sn(1), Sn(2), Sn(3) sites in the unit cell, we revealed that the unstable phonon modes are associated with the structural distortions involving the planar Sn(1) and Sc sites, while the contribution from the V kagome layer is negligible (Fig. 4e). This result not only explains our observation of the large charge order gap on the Sn(1) bands (Fig. 3) and the marginal change of the V kagome bands (Fig. 2), but also is fully consistent with the X-ray refined charge order structure of ScV6Sn6 that revealed the dominant distortions in the Sn(1) and Sc sites37. It is also instructive to compare the phonon modes of ScV6Sn6 to those of the CsV3Sb5 shown in Fig. 4f, g. In stark contrast to the case of ScV6Sn6, the unstable phonon modes of CsV3Sb5 at M and L (associated with the 2 × 2 charge order) accompany the dominant displacement of the V atoms, and reflect the intrinsic electronic instability from the V kagome layers (Supplementary Fig. 11)40.
Phase diagram of charge order in Sc(V1−xCrx)6Sn6
Finally, we construct the phase diagram of Sc(V1−xCrx)6Sn6 series to understand the evolution of charge order with carrier doping (Fig. 5). The charge order phase remains robust in the wide-doping range, up to doping x ≈ 0.10 charges per V atom. In the framework of the virtual crystal approximation (Supplementary Fig. 12), this indicates that the charge order phase remains stable up to the order of 120 meV Fermi level shift, further making the electronic scenarios sensitive to the Fermi level, such as the Fermi surface or vHS nesting, unlikely. We note that this response of the charge order to carrier doping is again highly different from CsV3Sb5, where the charge order rapidly vanishes after the x ≈ 0.02 ~ 0.03 charge doping per V atom, regardless of the doping methods31,41. Such a rapid suppression of charge order in CsV3Sb5 with a slight shift of the chemical potential (that disturbs the nesting conditions) is consistent with the electronic origin of the charge order in AV3Sb5. Meanwhile, our powder X-ray diffraction characterizations of the Sc(V1-xCrx)6Sn6 series revealed that Cr substitution mainly affects the c-axis lattice parameter (Supplementary Fig. 13). Given that the \(\sqrt{3}\)×\(\sqrt{3}\) order in ScV6Sn6 mainly accompanies the c-axis motion of the Sc and Sn(1) atoms37, we suspect that the reduction of the c-axis lattice parameter is the main driving force of the charge order suppression in the Sc(V1-xCrx)6Sn6 phase diagram.

a Evolution of the normalized resistivity T/T300K as a function of Cr-doping in x = 0, 0.01, 0.02, 0.03, and 0.06 samples. b Doping-temperature phase diagram of the charge order in ScV6Sn6, error bars indicate the range of resistance change during transitions.
In summary, the present work elucidates the origin of the \(\sqrt{3}\)×\(\sqrt{3}\) charge order in the newly discovered kagome metal ScV6Sn6. Our comprehensive characterizations of the electronic structure, phonon dispersion, and phase diagram coherently emphasize the essential role of the structural degrees of freedom other than the V kagome lattice in driving the charge order. In this context, the nature of the \(\sqrt{3}\)×\(\sqrt{3}\) charge order in ScV6Sn6 is fundamentally different from the 2×2 charge order in the archetype kagome metal AV3Sb5, where the electronic instability in the V kagome lattice plays a major role. As discussed in the introduction, the true charge disproportionation phases in the kagome lattice can support the exotica of physics, including the fractionalization of elementary particles. While ScV6Sn6 is not an ideal platform to serve this purpose, our study underscores that the search for new kagome quantum materials hosting various types of genuine charge orders should be continued.
Note added in proof: while preparing the manuscript, relevant works on the electronic structure and phonon dispersions of ScV6Sn6 were reported42,43,44,45,46. The initial ARPES and optical spectroscopy characterizations in Ref. 42,43,44 failed to identify the charge order gap. Our observation of the charge order gap in the Sn(1) band is consistent with the ARPES spectrum in ref. 45 The structural instability identified in our phonon calculation is consistent with the report of ref. 46.
Methods
Single crystal synthesis and characterization
Single crystals of ScV6Sn6 and Sc(V1−xCrx)6Sn6 doping series were grown by typical self-flux methods. Scandium pieces (99.9% Research Chemicals), Vanadium pieces (99.7% Alfa Aeser), and Sn ingot (99.99% Alfa Aeser) were put in the Alumina crucible with frit disc, then sealed in Ar-gas purged evacuated quartz tube. Ampule was heated at 1100 °C for 24 h, then slow cooled to 800 °C with 1 ~ 2 °C/h cooling ratio. To remove the flux, ampule was centrifuged at 800 °C. V and Cr ratios of the doping series were confirmed using energy dispersive spectroscopy. Electrical Resistivity measurements was performed with Physical Properties Measurement System (PPMS, Quantum design) using a conventional 4 probe method. The X-ray diffraction measurements were conducted using Cu Kα1 source (λ = 1.54 Å) and 6-axis diffractometer. We identified (1/3 1/3 19/3), (1/3 1/3 20/3), (2/3 2/3 19/3), and (2/3 2/3 20/3) peaks associated with the \(\sqrt{3}\)×\(\sqrt{3}\)×3 charge order, all displaying the same temperature-dependence.
ARPES experiments
ARPES experiments were conducted at Beamline 7.0.2 (MAESTRO) and Beamline 4.0.3 (MERLIN) of the Advanced Light Source, equipped with R4000 and R8000 hemispherical electron analysers (Scienta Omicron), respectively. The samples were cleaved inside ultra-high vacuum chambers with a base pressure better than 4 × 10−11 Torr. To identify high-symmetry planes of the three-dimensional bulk Brillouin zone of ScV6Sn6, the photon energy dependent ARPES measurements were performed in a wide phonon energy range from 60 eV to 200 eV. By comparing the experimental kz dispersion to the DFT band structure, we identified 129 eV and 115 eV photon energies measuring the kz ≈ 0 and kz ≈ π high-symmetry planes, respectively. The data in Fig. 2 are acquired with 129 eV photons, while the data in Fig. 3 are measured with 115 eV photons. All normal state (charge-ordered state) data in the main text is obtained at 120 K (6 K) using linear horizontal light polarization, unless specified. We refer to Supplementary Fig. 9 for the data measured in finer temperature steps.
Spatially resolved ARPES and XPS experiments
The real-space mappings of the valence band structure and core level spectra were conducted at Beamline 7.0.2 (MAESTRO) of the Advanced Light Source. To resolve the complex surface domains of ScV6Sn6, we used the micro-focused synchrotron of lateral dimension 30 × 30 μm2. The domain dependent ARPES and XPS spectra are compared to the slab DFT calculations of various geometries, to assign the atomic termination layer to each domain (Supplementary Note 2, 3).
DFT calculations
DFT calculations were performed using the Vienna AB initio Simulation Package software47,48. The generalized-gradient approximation Perdew-Burke-Ernzerhof exchange-correlation functional was chosen to calculate the exchange-correlation energy49. The pseudopotential was defined based on the projector augmented-wave method50. VASPKIT software was used for pre- and post-processing of DFT calculated information51. For the bulk band calculation of ScV6Sn6, we used the lattice parameters (a, b, c) = (5.456 Å, 5.456 Å, 9.230 Å) which is obtained by relaxing the reported single crystal refinement data37. Relaxation is performed at the 350 eV kinetic energy cutoff that fully covers the atomic energy. The static electronic structure was calculated using a Γ-centered k-point mesh, 15 × 15 × 8 for the normal state structure and 8 × 8 × 3 for the charge ordered structure. We present the overall band dispersion of ScV6Sn6 in the normal state with and without spin-orbit coupling in Supplementary Fig. 10a. Supplementary Fig. 10b displays the unfolded DFT band dispersion in the charge ordered state. To understand the termination-dependence of the valence band and core level spectra, we performed the slab DFT calculation on all possible charge neutral slab configurations of ScV6Sn6 relaxed at the 350 eV kinetic cutoff energy (Supplementary Note 2, 3). Γ-centered 11 × 11 × 1 k-point mesh were used for the slab band calculation. Each slab has 20 atomic layers and the vacuum was fixed at 20 Å.
DFPT calculations
Phonon dispersions were computed within the density functional perturbation theory (DFPT) framework. Input parameters were generated from the 3 × 3 × 2 supercell using Phonopy software52,53. We compared the phonon modes of two kagome metals, ScV6Sn6 and CsV3Sb5. Various smearing factors 0.10, 0.125, 0.15, 0.175, and 0.20 were tested for both compounds. We present the smearing factor-dependent phonon dispersions in Supplementary Fig. 11.
Data availability
Original data can be found at https://doi.org/10.7910/DVN/HRCVXB. Other data supporting the findings of this study are available from the corresponding author on reasonable request.
Code availability
The datasets presented within this study are available from the corresponding authors upon reasonable request.
References
Sato, M. & Ando, Y. Topological superconductors: a review. Rep. Prog. Phys. 80, 1–45 (2017).
Mielke, A. Exact ground states for the hubbard model on the kagome lattice. J. Phys. A. Math. Gen. 25, 4335–4345 (1992).
Kiesel, M. L., Platt, C. & Thomale, R. Unconventional fermi surface instabilities in the kagome Hubbard model. Phys. Rev. Lett. 110, 126405 (2013).
Kiesel, M. L. & Thomale, R. Sublattice interference in the kagome Hubbard model. Phys. Rev. B 86, 121105 (2012).
Yu, S. L. & Li, J. X. Chiral superconducting phase and chiral spin-density-wave phase in a Hubbard model on the kagome lattice. Phys. Rev. B 85, 144402 (2012).
Wang, W., Li, Z., Xiang, Y. & Wang, Q. Competing electronic orders on kagome lattices at van Hove filling. Phys. Rev. B 87, 115135 (2013).
Guo, H. M. & Franz, M. Topological insulator on the kagome lattice. Phys. Rev. B 80, 113102 (2009).
Xu, G., Lian, B. & Zhang, S.-C. Intrinsic quantum anomalous Hall effect in the kagome lattice Cs2LiMn3F12. Phys. Rev. Lett. 115, 186802 (2015).
Tang, E., Mei, J.-W. & Wen, X.-G. High-temperature fractional quantum Hall states. Phys. Rev. Lett. 106, 236802 (2011).
Nakatsuji, S., Kiyohara, N. & Higo, T. Large anomalous Hall effect in a non-collinear antiferromagnet at room temperature. Nature 527, 212–215 (2015).
Ye, L. et al. Massive Dirac fermions in a ferromagnetic kagome metal. Nature 555, 638–642 (2018).
Liu, E. et al. Giant anomalous Hall effect in a ferromagnetic kagome-lattice semimetal. Nat. Phys. 14, 1125–1131 (2018).
Liu, D. F. et al. Magnetic weyl semimetal phase in a Kagomé Crystal. Science 365, 1282–1285 (2019).
Kang, M. et al. Dirac fermions and flat bands in the ideal kagome metal FeSn. Nat. Mater. 19, 163–169 (2020).
Kang, M. et al. Topological flat bands in frustrated kagome lattice CoSn. Nat. Commun. 11, 4004 (2020).
Yin, J. X. et al. Quantum-limit Chern topological magnetism in TbMn6Sn6. Nature 583, 533–536 (2020).
Ortiz, B. R. et al. CsV3Sb5: a Z2 topological kagome metal with a superconducting ground state. Phys. Rev. Lett. 125, 247002 (2020).
Zhao, H. et al. Cascade of correlated electron states in a kagome superconductor CsV3Sb5. Nature 599, 216–221 (2021).
Nie, L. et al. Charge-density-wave-driven electronic nematicity in a kagome superconductor. Nature 604, 59–64 (2022).
Chen, H. et al. Roton pair density wave and unconventional strong-coupling superconductivity in a topological kagome metal. Nature 599, 222–228 (2021).
Kang, M. et al. Twofold van Hove singularity and origin of charge order in topological kagome superconductor CsV3Sb5. Nat. Phys. 18, 301–308 (2022).
Cho, S. et al. Emergence of new van Hove singularities in the charge density wave state of a topological kagome metal RbV3Sb5. Phys. Rev. Lett. 127, 236401 (2021).
Wu, X. et al. Nature of unconventional pairing in the kagome superconductors AV3Sb5 (A = K, Rb, Cs). Phys. Rev. Lett. 127, 177001 (2021).
Feng, X., Jiang, K., Wang, Z. & Hu, J. Chiral flux phase in the kagome superconductor AV3Sb5. Sci. Bull. 66, 1384–1388 (2021).
Denner, M. M., Thomale, R. & Neupert, T. Analysis of charge order in the kagome metal AV3Sb5 (A=K,Rb,Cs). Phys. Rev. Lett. 127, 217601 (2021).
Jiang, Y.-X. et al. Unconventional chiral charge order in kagome superconductor KV3Sb5. Nat. Mater. 20, 1353–1357 (2021).
Mielke, C. et al. Time-reversal symmetry-breaking charge order in a kagome superconductor. Nature 602, 245–250 (2022).
Yang, S. Y. et al. Giant, unconventional anomalous Hall effect in the metallic frustrated magnet candidate, KV3Sb5. Sci. Adv. 6, eabb6003 (2020).
Kang, M. et al. Charge order landscape and competition with superconductivity in kagome metals. Nat. Mater. 22, 186–193 (2023).
Chen, K. Y. et al. Double superconducting dome and triple enhancement of Tc in the kagome superconductor CsV3Sb5 under high pressure. Phys. Rev. Lett. 126, 247001 (2021).
Oey, Y. M. et al. Fermi level tuning and double-dome superconductivity in the kagome metals CsV3Sb5-xSnx. Phys. Rev. Mater. 6, L041801 (2022).
Wen, J., Rüegg, A., Wang, C. C. J. & Fiete, G. A. Interaction-driven topological insulators on the kagome and the decorated honeycomb lattices. Phys. Rev. B 82, 075125 (2010).
Rüegg, A. & Fiete, G. A. Fractionally charged topological point defects on the kagome lattice. Phys. Rev. B 83, 165118 (2011).
Nishimoto, S., Nakamura, M., O’Brien, A. & Fulde, P. Metal-insulator transition of fermions on a kagome lattice at 1/3 filling. Phys. Rev. Lett. 104, 196401 (2010).
Ferrari, F., Becca, F. & Valentí, R. Charge-density waves in kagome-lattice extended Hubbard models at the van Hove filling. Phys. Rev. B 106, L081107 (2022).
Han, T. H. et al. Fractionalized excitations in the spin-liquid state of a kagome-lattice antiferromagnet. Nature 492, 406–410 (2012).
Arachchige, H. W. S. et al. Charge density wave in kagome lattice intermetallic ScV6Sn6. Phys. Rev. Lett. 129, 216402 (2022).
Zhou, X. et al. Origin of charge density wave in the kagome metal CsV3Sb5 as revealed by optical spectroscopy. Phys. Rev. B 104, L041101 (2021).
Liu, Z. et al. Charge-density-wave-induced bands renormalization and energy gaps in a kagome superconductor RbV3Sb5. Phys. Rev. X 11, 41010 (2021).
Christensen, M. H., Birol, T., Andersen, B. M. & Fernandes, R. M. Theory of the charge-density wave in AV3Sb5 kagome metals. Phys. Rev. B 104, 214513 (2021).
Yang, H. et al. Titanium doped kagome superconductor CsV3-xTixSb5 and two distinct phases. Sci. Bull. 67, 2176–2185 (2022).
Hu, T. et al. Optical studies of structural phase transition in the vanadium-based kagome metal ScV6Sn6. Phys. Rev. B 107, 165119 (2023).
Tuniz, M. et al. Dynamics and resilience of the unconventional charge density wave in ScV6Sn6 bilayer kagome metal. Commun. Mater. 4, 103 (2023).
Cheng, S. et al. Nanoscale visualization and spectral fingerprints of the charge order in ScV6Sn6 distinct from other kagome metals. Preprint at https://doi.org/10.48550/arXiv.2302.12227 (2023).
Korshunov, A. et al. Softening of a flat phonon mode in the kagome ScV6Sn6. Nat. Commun. 14, 6646 (2023).
Tan, H. & Yan, B. Abundant lattice instability in kagome metal ScV6Sn6. Phys. Rev. Lett. 130, 266402 (2023).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Blochl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Wang, V., Xu, N., Liu, J.-C., Tang, G. & Geng, W.-T. VASPKIT: A user-friendly interface facilitating high-throughput computing and analysis using VASP code. Comput. Phys. Commun. 267, 108033 (2021).
Togo, A. & Tanaka, I. First principles phonon calculations in materials science. Scr. Mater. 108, 1–5 (2015).
Togo, A. First-principles Phonon Calculations with Phonopy and Phono3py. J. Phys. Soc. Jpn. 92, 12001 (2022).
Acknowledgements
The works at Max Planck POSTECH/Korea Research Initiative were supported by the National Research Foundation of Korea funded by the Ministry of Science and ICT, Grant No. 2022M3H4A1A04074153 and 2020M3H4A2084417. This research used resources of the Advanced Light Source, a U.S. DOE Office of Science User Facility under contract no. DE-AC02-05CH11231. R.C. acknowledges the support from the Air Force Office of Scientific Research grant FA9550-22-1-043, and the STC Center for Integrated Quantum Materials, NSF grant DMR-1231319. M.K. acknowledges a Samsung Scholarship from the Samsung Foundation of Culture. This paper is supported by Basic Science Research Institute Fund, whose NRF grant number is 2021R1A6A1A10042944.
Author information
Authors and Affiliations
Contributions
R.C., M.K., and J.-H.P. conceived the project; S.L., J.K., J.Y., and S.P. performed the ARPES experiments and analyzed the resulting data with help from J.D., C.J., A.B., and E.L.; S.L. performed the theoretical calculations; C.W. synthesized and characterized the crystals.; S.L. and M.K. wrote the manuscript with input from all coauthors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lee, S., Won, C., Kim, J. et al. Nature of charge density wave in kagome metal ScV6Sn6. npj Quantum Mater. 9, 15 (2024). https://doi.org/10.1038/s41535-024-00620-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41535-024-00620-y
