
Abstract
Congenital muscular dystrophies (CMDs) are a group of rare muscle disorders characterized by early onset hypotonia and motor developmental delay associated with brain malformations with or without eye anomalies in the most severe cases. In this study, we aimed to uncover the genetic basis of severe CMD in Egypt and to determine the efficacy of whole exome sequencing (WES)-based genetic diagnosis in this population. We recruited twelve individuals from eleven families with a clinical diagnosis of CMD with brain malformations that fell into two groups: seven patients with suspected dystroglycanopathy and five patients with suspected merosin-deficient CMD. WES was analyzed by variant filtering using multiple approaches including splicing and copy number variant (CNV) analysis. We identified likely pathogenic variants in FKRP in two cases and variants in POMT1, POMK, and B3GALNT2 in three individuals. All individuals with merosin-deficient CMD had truncating variants in LAMA2. Further analysis in one of the two unsolved cases showed a homozygous protein-truncating variant in Feline Leukemia Virus subgroup C Receptor 1 (FLVCR1). FLVCR1 loss of function has never been previously reported. Yet, loss of function of its paralog, FLVCR2, causes lethal hydranencephaly-hydrocephaly syndrome (Fowler Syndrome) which should be considered in the differential diagnosis for dystroglycanopathy. Overall, we reached a diagnostic rate of 86% (6/7) for dystroglycanopathies and 100% (5/5) for merosinopathy. In conclusion, our results provide further evidence that WES is an important diagnostic method in CMD in developing countries to improve the diagnostic rate, management plan, and genetic counseling for these disorders.
Similar content being viewed by others


Introduction
Congenital muscular dystrophies (CMDs) are a clinically and genetically heterogeneous group of disorders characterized by hypotonia within the first year of life, motor development delay, and progressive muscle weakness in addition to dystrophic features on muscle biopsy and elevated creatine kinase (CK) [1]. Brain malformations, with or without accompanying eye anomalies, are key features of severe forms of CMD such as the dystroglycanopathies [including Muscle-Eye-Brain (MEB), Fukuyama CMD (FCMD) and Walker-Warburg syndrome (WWS)] and merosinopathies (merosin-deficient CMD).
To date, mutations in 18 genes have been reported to cause dystroglycanopathies, each of which are involved in glycosylation of α-dystroglycan (α- DG) [2], while merosin-deficient CMD is caused by mutations in the laminin-α2 gene (LAMA2), a component of the laminin subtype, merosin [3]. Together, these genes functionally converge on the regulation of interactions between cells and the basement membrane and extracellular matrix (ECM), as the functional glycan on α-DG binds ECM ligands such as merosin and other laminin G-like domain-containing proteins [4].
The worldwide prevalence of all CMDs ranges from 0.5 to 0.9 per 100,000 [5, 6]. In Egypt, one study in 2005 reported that the prevalence of combined muscular dystrophies was 26.8 per 100,000, while CMD prevalence was as high as 3.8 per 100,000 [7]. The prevalence of more severe forms of dystroglycanopathy with brain and eye malformations (MEB, FCMD, and WWS) are more difficult to quantify due to their early lethality. Studies focused on geographical distribution of mutations in severe dystroglycanopathy have been helpful in defining the most common genetic causes. European cohorts have identified mutations in POMT1 as the most frequent cause of WWS [5, 8], but this also held true in an ethnically diverse cohort [8]. In contrast, POMGNT1 mutations are the most frequent cause of MEB overall [9]. However, a founder retrotransposon insertion in the 3’ UTR of FKTN contributes to the majority of CMDs in Japan [10], and this same insertion was also identified in 14/42 dystroglycanopathy patients in a South Korean cohort [11]. Similarly, a different founder mutation in FKTN with a carrier frequency of 0.7% leads to WWS in the Ashkenazi Jewish population [12]. Merosinopathy is the most common CMD in the United Kingdom [5], the 2nd and the 3rd most common cause in Italy and Australia respectively [6, 13]. However, it accounts for only 6% of CMD in Japan [14]. As such, regional differences can be expected.
Next-generation sequencing (NGS) technologies such as whole-exome sequencing (WES) or gene panels have had a great impact on the medical diagnosis of CMDs, increasing the speed of genetic diagnosis in developed countries [15] and even in developing countries according to recent studies in Africa and Jordan [16, 17]. As multiple drug and gene therapy-based approaches are being approved or tested for other forms of muscular dystrophy, it is imperative to define the genetic distribution of CMD mutations in different populations which could benefit from future therapies.
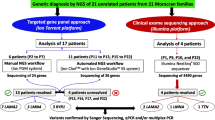
Despite global progress, CMDs remain understudied in developing countries. To address this gap, we recruited 12 patients from 11 Egyptian families presenting with CMDs with brain involvement. The overarching goal of this study is to provide a genetic diagnosis for each patient, which can improve traditional methods of diagnosis and genetic counseling, as well as treatment and research strategies.
Materials and methods
Subjects
Ethics approval was received from the Ethics Committee of Medical Research Institute and Faculty of Medicine, Alexandria University and the Institutional Review Board at Rutgers University. All patients/guardians were fully informed of the purpose and procedures of this study and written consent was obtained before participation. A full clinical assessment, pedigree, and family history for all patients was completed at the Human Genetic Clinic, Medical Research Institute in Alexandria, Egypt.
Whole exome sequencing and analysis
Genomic DNA was extracted from blood samples using a QIAmp DNA Mini Kit (Qiagen). Library preparation and whole exome sequencing was performed by Novogene Corporation Inc. (Sacramento, CA) using a SureSelect Human All Exon V6 kit (Agilent) and NovaSeq 6000 sequencer. Sequencing reads were mapped to hg38 with BWA [18], sorted with Sambamba [19], and merged with Picard, and variants were called with GATK [20], as part of Novogene’s Bioinformatics Analysis pipeline. Variant call format (VCF) files were then annotated with ANNOVAR [21], stored in a custom SQL database, and filtered for variants with allele frequencies < 1% in gnomAD v3.1.2 and the Greater Middle Eastern Variome (GME) Northeast Africa and Arabian Peninsula populations [22]. Pathogenicity predictors including SIFT [23], PolyPhen2 [24], and CADD [25] were used to assess the effect of missense variants, and SpliceAI [26] was used to evaluate intronic and synonymous variants for their potential to disrupt splicing. In addition, ExomeDepth [27] was used to identify any copy number variants (CNVs) affecting genes known to cause CMD using unrelated individuals as controls.
Results
Clinical description
All individuals in our cohort presented with early onset hypotonia, brain abnormalities in magnetic resonance imaging (MRI), myopathic changes on EMG, and/or elevated serum creatinine kinase suggestive of CMD. However, the overall clinical and radiological presentation defined 2 groups. Group I included seven patients (four females and three males) with suspected dystroglycanopathy, and Group II was comprised of five patients (one female and four males) with suspected merosin-deficient CMD (Table 1). Clinical assessment alone was not enough for a definitive diagnosis in S7, however, as he exhibited manifestations of both MEB and merosin-deficient CMD.
All of the cases in Group I and II were born from consanguineous unions, apart from S2. However, both parents of S2 originated from the same village. The age at the time of the first visit to the genetic clinic was significantly younger in Group I (1 day to 7 months old) than Group II (2 to 7 years old). Several cases had a family history suggestive of CMDs. For instance, S1 had a distant cousin with hydrocephalus (Fig. 1A), S3 had one deceased sib with hydrocephalus and failure to thrive (Fig. 1B), S5 had two siblings who died during the neonatal period suffering from severe hypotonia (Fig. 1C), and S7 had a distant relative with late onset muscle disease. Among cases of Group II, S11 and S12 are siblings with a previous history of an older sibling with the same condition who died at age of 9 (Fig. 1D).

Pedigrees for cases with family history of CMD. A S1 was born from a 1st cousin union and had a distant cousin diagnosed with hydrocephalus. B S3 was born from a 1st cousin union and had a deceased younger brother with hydrocephalus and failure to thrive. C S5 was born from a 1st cousin union and had two siblings with severe hypotonia who died in infancy. D S11 and S12 are siblings and had an older sib with CMD who died at age 9
Except for S7, individuals in Group I exhibited severe global developmental delay with no motor development present in S1, S2, and S6 (Table 1). They had no head control and failed to reach any motor milestones. Most individuals had highly elevated CK levels and myopathic changes were seen on EMG. No muscle biopsies were conducted. Additionally, each individual in Group I had absent speech, eye abnormalities ranging from ptosis to microphthalmia and retinal detachment, and five of them presented with seizures that were controlled by anti-epileptic drugs (AEDs). Four patients (S3-6) died before the age of two. Interestingly, S7 presented with milder clinical manifestations compared to the rest of Group I. During the last follow-up at 30 months of age, he showed typical age-appropriate behaviors, including responding to his name and following simple instructions, but he was unable to speak. The maximum motor achievement was sitting without support by age 1. He had highly elevated CK and only mild ptosis in his left eye.
Individuals in Group II achieved further milestones than individuals in Group I. Although none had achieved independent ambulation, four out of the five individuals (S8, S9, S11, S12) were able to sit without support by the age of two. S8 learned to write despite the presence of contractures and S10 only had neck support. Contractures were present in 3 patients (S8, S10, S11) and were progressive, affecting mainly knees and elbows. CK levels were not as elevated compared to Group I, but all showed myopathic changes upon EMG analysis. In addition, all individuals in Group II had normal cognitive function and behavior and none had seizures or ocular abnormalities. S8 and S10 had normal language development, while S9 only started to speak at 3 years of age and the siblings S11 and S12 said the first words at 2 years of age and had a mild deficit in articulation despite an IQ result in the typical range.
Radiological analysis of brain abnormalities
The findings of the brain MRI further supported the suspected diagnoses. All individuals in Group II showed the classical abnormal white matter myelination manifested as symmetrical T2 hyperintense signal in the periventricular and deep white matter that is typically observed in merosin-deficient CMD (Fig. 2A). Brain MRI for S8 also showed additional cortical malformations including bilateral occipital lissencephaly (Fig. 2B–C).

Brain imaging for selected cases. A Axial T2 MRI image showing the classical T2 hyperintense signal within the periventricular white matter in case S9. B, C Axial T2 MRI images of case S8 showing abnormal hyperintense signal within the periventricular white matter with additional cortical malformations including bilateral occipital segmental lissencephaly (arrows in C). D, E: Axial (D) and sagittal (E) T2 MRI images of case S1 showing diffuse cobblestone type II lissencephaly with flattening of the entire cerebral mantle as well as hypoplastic cerebellum and pons and Z-shaped brainstem (arrow in E). F, G: Different axial T2 MRI images of case S2 showing supratentorial abnormal perisylvian cortical development (arrows in F), cerebellar hypoplasia, small size pons, and abnormal right eye globe with abnormal right lens configuration and intra vitreous hemorrhage (asterisk in G). H, I: Coronal (H) and sagittal (I) non contrast CT images of case S5 showing the supratentorial brain nearly replaced by CSF apart from the interhemispheric fissure, also associated with hypoplastic cerebellum and kinked brain stem structures. J, K: Axial T2 MRI images of case S7 showing periventricular white matter abnormalities in addition to hypoplasia of both cerebellar hemispheres with multiple bilateral microcysts as well as dysplastic features
In contrast, Group I had a more variable presentation in the WWS/MEB spectrum. Brain MRI for S1, S4, and S6 showed lissencephaly with cobblestone malformation with flattening of the entire cerebral mantle as well as hypoplastic cerebellar hemispheres, cerebellar vermis, and z-shaped hypoplastic pons (Fig. 2D–E). S2 had supratentorial ventricular dilatation, abnormal perisylvian cortical development, cerebellar hypoplasia, microcysts, small size pons, and hypertrophied tectal plates (Fig. 2F–G). Only non-contrast Computed Tomography (CT) imaging was available for S5 showing the supratentorial brain nearly replaced by CSF apart from the interhemispheric fissure, and hypoplastic cerebellum and brain stem structures (Fig. 2H–I). S7 had white matter abnormalities in addition to hypoplasia of both cerebellar hemispheres with multiple bilateral microcysts as well as dysplastic features (Fig. 2J–K).
Whole exome sequencing analysis
DNA from all affected individuals and their parents was sent for WES. Initial analysis of known CMD genes reached a genetic diagnosis in 71% (5/7) of Group I with two homozygous variants in FKRP and one homozygous variant each in POMT1, POMK, and B3GALNT2. In Group II, a diagnosis was reached for 100% (5/5) of cases, as all had homozygous truncating variants in LAMA2 (Table 2). All parents were confirmed to be heterozygous carriers apart from S6 where parental DNA was not available.
S1 had a homozygous missense variant in POMK (NM_032237.5:c.641A > T; NP_115613.1:p.Gln214Leu). This variant is not present in population databases (gnomAD, GME), never reported in literature, and highly conserved among different species. Algorithms developed to predict the effect of missense changes on protein structure and function (SIFT, PolyPhen-2, CADD) suggest that this variant is likely to be disruptive. S2 had 2 bp frameshift deletion in POMT1 (NM_007171.4:c.2179_2180del; NP_009102.4:p.Ser727fs) which creates a premature stop codon and has been previously reported in two other cases [28]. S3 had a stop-gain variant in FKRP (NM_024301.5:c.778G > T; NP_077277.1:p.Glu260Ter) which is absent in gnomAD and GME. This variant has been only reported 3 times in ClinVar. S4 had a stop-gain variant in B3GALNT2 (NM_152490.5:c.1338G > A; NP_689703.1:pTrp446Ter) which is not found in gnomAD, GME, but is present in one individual in ClinVar. S7 had a missense variant in FKRP (NM_024301.5:c.1364C > A; NP_077277.1:p.Ala455Asp) that has been previously reported in the Tunisian, Moroccan, and Arab populations [29,30,31].
In the merosin-deficient CMD group, WES identified 4 different homozygous variants in LAMA2. S8 had a homozygous stop-gain variant (NM_000426.4:c.6955C > T; NP_000417.3:p.Arg2319Ter) that has been reported twice in trans with other LAMA2 variants [32, 33]. S9 had a homozygous stop-gain LAMA2 (NM_000426.4:c.4645C > T; NP_000417.3:p.Arg1549Ter) which has been reported in several studies [34,35,36]. S10 had a novel 4 bp deletion in LAMA2 (NM_000426.4:c.3886_3889del; NP_000417.3:p.Ile1296AlafsTer), and the siblings S11 and S12 had a novel 2 bp frameshift deletion in LAMA2 that lead to premature stop codon (NM_000426.4:c.6636_6637del; NP_000417.3:pGly2213SerTer).
Following WES analysis of protein-altering variants, cryptic splicing analysis, and CNV prediction, only two cases, S5 and S6, remained genetically unsolved. In S5, five candidate genes were left following rare variants filtering (PIP5K1C, PRX, HRC, ADAT3, and FLVCR1). Among these, the only protein-truncating variant was a novel homozygous frameshift insertion in Feline Leukemia Virus subgroup C Receptor 1 (FLVCR1; NM_014053:c.215dupC; NP_054772.1:p.E74Rfs). In S6, coverage-based CNV prediction with ExomeDepth predicted a CNV encompassing POMK in addition to several of its inferred enhancers listed in the GeneHancer database [37]. However, we are unable to confirm whether this predicted duplication is de novo or inherited, and further experiments would be required to confirm its presence and determine its effect on POMK expression. We also identified rare, homozygous variants in four possible candidate genes (DCTN2, DNAH5, GBE1, and NEURL1). Of interest was a homozygous missense variant in NEURL1 (NM_004210.5:c.1051G > T; NP_004201.3:p.Gly351Cys). This variant was absent in gnomAD and GME, occurred at a conserved amino acid, and is predicted to be damaging by SIFT (0.002), PolyPhen2 [1], and CADD [27]. Overall, we identified variants in 86% (6/7) of individuals in Group I and 100% (5/5) of individuals in Group II, with candidate variants in S6 where further investigation is warranted.
Discussion
In this study, we present one of the first cohorts from Egypt comprised of patients with CMD and brain malformations. Each patient underwent a comprehensive clinical workup and were grouped as those with suspected dystroglycanopathy (Group I) or suspected merosin-deficient CMD (Group II). However, this workup is not always sufficient to provide a definitive diagnosis, especially given the clinical heterogeneity of brain malformations observed in severe CMDs. As such, we leveraged WES to identify variants causing dystroglycanopathies and merosin-deficient CMD, several of which were novel or reported for the first time in Egypt. Overall, WES represents a powerful technology for detecting disease causing variants.
The diagnostic rate achieved in this study was very high with a potentially disease-causing mutation detected in 86% (6/7) of individuals in Group I (suspected dystroglycanopathy) and 100% (5/5) in Group II (suspected merosin-deficient CMD). All variants identified in this study were found in homozygosity despite only 9/12 enrolled probands being born from first cousin (consanguineous) parents. It is estimated that about 30% of all marriages in Egypt in the last 40 years have been consanguineous [38]. Hence, our data emphasizes the higher burden of severe, rare diseases in consanguineous populations since referral to our center or recruitment in this study was not restricted to consanguineous unions. The lack of a national registry system for rare diseases along with the insufficiency of diagnostic tools makes it difficult to reach the precise rate of rare disorders in Egypt. However, a large study conducted at one genetic center at Cairo University in the period between 1966 to 2009 showed that the frequency of genetic disorders was 4.3% [39]. In addition, about 23,000 families with known or suspected genetic disorders were registered in the genetics clinics at the Medical Research Institute since 1981, and several studies of specific genetic disorders were conducted to underline not only the frequency but also the genetic makeup of these syndromes in Egypt [40, 41].
One of the goals of our study was to define whether common founder mutations in specific genes for dystroglycanopathies were present in the Egyptian population. The number of studies that have focused exclusively on this type of CMD is limited with diverse population-specific variants being identified. For example, a Middle Eastern founder mutation in LAMA2 (c.3924 + 2 T > C) that causes an in-frame deletion of 63 amino acids was identified in four families from Saudi Arabia and one family from Sudan [42, 43]. This variant likely originated in Saudi Arabia and later entered North Africa, which may be relevant to individuals with merosin-deficient CMD in Egypt [42, 44]. Furthermore, a founder FKTN variant accounts for the majority of severe CMD (FCMD) in Japan, China, and South Korea [10, 11, 45], and a different founder mutation in FKTN leads to WWS in Ashkenazi Jews [8, 12]. In contrast, POMT1 and POMGNT1 account for 30–40% of MEB/WWS cases in European populations [8]. Among the dystroglycanopathy cohort, we detected pathogenic variants in 4 different genes in 5 cases. The only gene that was found mutated twice was FKRP with two different variants causing variable clinical manifestations: S3 harbored a novel stop-gain variant and had WWS with cerebellar microcysts and hydrocephalus, while S7 harbored a missense variant and presented with a phenotype overlapping MEB and merosin-deficient CMD characterized by white matter hyperintensities, cerebellar hypoplasia, and microcysts without hydrocephalus or cortical malformation.
The missense variant in FKRP identified in S7 (p.Ala455Asp) was previously reported as a founder mutation in the Tunisian population and was also reported in other families of Arabian descent [29,30,31]. This is the only Arab/North African founder mutation identified in our cohort. Further, this variant is associated with a variable clinical presentation [30]. Our case differs from previous cases with severe developmental delay in having appropriate behavioral responses for his age despite the lack of language. This could suggest the presence of unidentified modifiers of FKRP.
The remaining three genetically confirmed dystroglycanopathy cases each had variants in different genes. While variants in POMT1 are the most frequent cause of WWS in other populations, we only found one previously reported POMT1 frameshift variant in a single patient with a presentation consistent with MEB (S2). This variant also leads to variable presentation with two previous cases diagnosed with WWS and MEB [28]. Both presented with microcephaly like S2, but also had contractures which were absent in S2. The brain MRI abnormalities in the previous MEB case were similar to S2: hydrocephalus, cerebral hypoplasia with cerebral microcysts, in addition to brain stem abnormalities. In one of the WWS cases (S1), we identified a missense variant in POMK, which is a very rare cause of dystroglycanopathy. To date, only nine pathogenic POMK variants have been reported worldwide in 16 cases, and five of them were stillbirth or fetuses from induced termination of pregnancy [46]. Six out of nine variants cause WWS while the remaining three cause limb-girdle muscular dystrophy [46]. POMK genotype/phenotype correlations are complex because even variants leading to expression of a significantly truncated protein can result in a mild phenotype [47]. However, the functional and physiological mechanisms underlying the phenotypic variability remain unclear. We also identified a novel homozygous nonsense variant disrupting the glycosyltransferase domain of B3GALNT2 leading to WWS. B3GALNT2 variants also lead to variable presentations with 23 cases documented in the literature: 5 having WWS, 10 having MEB, and 8 with atypical CMD [48].
In S5, we identified a likely pathogenic variant in FLVCR1, which is not a known cause of dystroglycanopathy. FLVCR1 was shown to act as a heme transporter and was recently proposed to be a major choline transporter [49]. It is widely expressed but is most abundant in the retina, spinal cord, and brain, especially in the cerebellum and hippocampus [50]. Missense variants in FLVCR1 cause posterior column ataxia with retinitis pigmentosa [50], and homozygous protein-truncating variants have never been reported, though removal of Flvcr1 in mice is embryonic lethal [49]. However, loss of function variants of its paralog FLVCR2 cause a syndrome characterized by proliferative vasculopathy and hydranencephaly-hydrocephaly, also termed Fowler Syndrome [51]. Eye abnormalities, joint contractures, and muscle atrophy are also common in Fowler Syndrome. S5 presented with profound hydrocephaly, contractures, seizures, and optic nerve atrophy which would also be consistent with this syndrome. Since only a CT was available for this case, only severe hydrocephalus was ascertained leading to the possible dystroglycanopathy diagnosis. In addition, there is no autopsy information to confirm the distinctive vasculopathy. Considering the clinical overlap, it is likely that homozygous loss of function variants in FLVCR1 also cause a presentation consistent with Fowler Syndrome.
S6 was the only case in Group I that remained unsolved after a comprehensive analysis of rare variants. Among the variants identified, one potential candidate is NEURL1, which encodes an E3 ubiquitin ligase that plays a role in the regulation of Notch pathway and is expressed at variable levels throughout the muscle and brain [52]. Multiple regulators of the Notch pathway have been involved in muscle disease (MEGF10, POGLUT1, and JAG2) [53]. Furthermore, a unique variant in JAG1, which is not directly related to skeletal muscle disease, has been found to possess a modifying effect on muscular dystrophy [54], and NEURL1 had been found to affect the signaling activity of JAG1 by directly enhancing its ubiquitination [55]. Further investigations may be warranted to determine whether NEURL1 mutations could lead to dystroglycanopathy phenotypes. Additionally, we used ExomeDepth to predict CNVs from WES coverage data, as pathogenic CNVs have been identified in individuals with dystroglycanopathy in several studies including a ~ 63 kb intragenic deletion in LARGE1 in a case with WWS [56] and a ~ 1.6 kb deletion in POMGNT1 in an individual with MEB [57]. This strategy indicated that S6 may harbor a heterozygous duplication encompassing POMK and several of its inferred enhancers. While experimental assays have shown that ExomeDepth CNV predictions are often accurate [58], further analysis is warranted to confirm the presence of this CNV and evaluate its effect on POMK expression.
In Group II (suspected merosinopathy), we identified 4 different homozygous truncating variants in LAMA2: two stop-gain, and two frameshift deletions. The two frameshift deletions were novel, and the stop-gain variants p.Arg2319Ter and p.Arg1549Ter were reported eight and 14 times in the LOVD database, respectively (https://databases.lovd.nl/shared/genes/LAMA2). The p.Arg2319Ter mutation was associated with bilateral occipital pachygyria in addition to classical white matter changes, but this type of cortical malformation is not uncommon for individuals with merosin deficiency [4, 39]. In addition to S8, this variant was reported twice in trans with other variants: once with a deletion of exon 56 (c.7750_7899del) in a patient with hypotonia and severe dystrophic changes in muscle biopsy [32], and once with p.Cys2909Arg in a patient with late onset merosin-deficient CMD [33]. The variant identified in S9 (p.Arg1549Ter) has been reported several times in other studies with profound clinical variability. One patient with complete merosin deficiency was 17 years old and sitting without support, which was his maximum motor achievement [34]. Another patient presented with only partial merosin deficiency. They achieved ambulation with aid and were able to climb stairs with support [35]. Lastly, this mutation was detected in a compound heterozygosity in a patient with CMD, dilated cardiomyopathy, and life-threatening ventricular arrhythmias [36]. Overall, none of the patients in Group II could achieve independent walking, consistent with the findings that all the patients who carried biallelic premature termination codons never walk [3].
As the rate of genetic diagnosis for merosin-deficient CMD in our study is 100%, which has also been achieved in another study [59], we recommend that following clinical and radiological assessment, LAMA2 gene sequencing should be performed first in similar cases. However, availability and cost effectiveness remain two major concerns. For the dystroglycanopathies, we found mutations in 86% of cases in this study, but future studies on larger cohorts may benefit from additional NGS assays such as whole genome sequencing with long-read strategies, as we suspect noncoding variants that go undetected in WES may contribute to many unsolved cases.
Data availability
Data reported in this study will be made available upon request from the corresponding author.
References
Kang PB, Morrison L, Iannaccone ST, Graham RJ, Bonnemann CG, Rutkowski A et al (2015) Evidence-based guideline summary: evaluation, diagnosis, and management of congenital muscular dystrophy: Report of the guideline development subcommittee of the american academy of neurology and the practice issues review panel of the american association of neuromuscular & electrodiagnostic medicine. Neurology 84(13):1369–1378
Kanagawa M (2021) Dystroglycanopathy: From Elucidation of Molecular and Pathological Mechanisms to Development of Treatment Methods. Int J Mol Sci 22(23):13162
Oliveira J, Gruber A, Cardoso M, Taipa R, Fineza I, Goncalves A et al (2018) LAMA2 gene mutation update: Toward a more comprehensive picture of the laminin-alpha2 variome and its related phenotypes. Hum Mutat 39(10):1314–1337
Bouchet-Séraphin C, Vuillaumier-Barrot S, Seta N (2015) Dystroglycanopathies: About Numerous Genes Involved in Glycosylation of One Single Glycoprotein. J Neuromuscul Dis 2(1):27–38
Sframeli M, Sarkozy A, Bertoli M, Astrea G, Hudson J, Scoto M et al (2017) Congenital muscular dystrophies in the UK population: Clinical and molecular spectrum of a large cohort diagnosed over a 12-year period. Neuromuscul Disord 27(9):793–803
Graziano A, Bianco F, D’Amico A, Moroni I, Messina S, Bruno C et al (2015) Prevalence of congenital muscular dystrophy in Italy: a population study. Neurology 84(9):904–911
El-Tallawy HN, Khedr EM, Qayed MH, Helliwell TR, Kamel NF (2005) Epidemiological study of muscular disorders in Assiut, Egypt. Neuroepidemiology 25(4):205–211
Manzini MC, Gleason D, Chang BS, Hill RS, Barry BJ, Partlow JN et al (2008) Ethnically diverse causes of Walker-Warburg syndrome (WWS): FCMD mutations are a more common cause of WWS outside of the Middle East. Hum Mutat 29(11):E231–E241
Song D, Dai Y, Chen X, Fu X, Chang X, Wang N et al (2021) Genetic variations and clinical spectrum of dystroglycanopathy in a large cohort of Chinese patients. Clin Genet 99(3):384–395
Kondo-Iida E, Kobayashi K, Watanabe M, Sasaki J, Kumagai T, Koide H et al (1999) Novel mutations and genotype-phenotype relationships in 107 families with Fukuyama-type congenital muscular dystrophy (FCMD). Hum Mol Genet 8(12):2303–2309
Ko YJ, Cho A, Kim WJ, Kim SY, Lim BC, Kim H et al (2023) Broad spectrum of phenotype and genotype in Korean α-dystroglycan related muscular dystrophy presenting to a tertiary pediatric neuromuscular center. Neuromuscul Disord 33(5):425–431
Chang W, Winder TL, LeDuc CA, Simpson LL, Millar WS, Dungan J et al (2009) Founder Fukutin mutation causes Walker-Warburg syndrome in four Ashkenazi Jewish families. Prenat Diagn 29(6):560–569
O’Grady GL, Lek M, Lamande SR, Waddell L, Oates EC, Punetha J et al (2016) Diagnosis and etiology of congenital muscular dystrophy: We are halfway there. Ann Neurol 80(1):101–111
Allamand V, Guicheney P (2002) Merosin-deficient congenital muscular dystrophy, autosomal recessive (MDC1A, MIM#156225, LAMA2 gene coding for alpha2 chain of laminin). Eur J Hum Genet 10(2):91–94
Lavelle TA, Feng X, Keisler M, Cohen JT, Neumann PJ, Prichard D et al (2022) Cost-effectiveness of exome and genome sequencing for children with rare and undiagnosed conditions. Genet Med 24(11):2415–2417
Wiener EK, Buchanan J, Krause A, Lombard Z (2023) Retrospective file review shows limited genetic services fails most patients - an argument for the implementation of exome sequencing as a first-tier test in resource-constraint settings. Orphanet J Rare Dis 18(1):81
Ababneh NA, Ali D, Al-Kurdi B, Barham R, Bsisu IK, Dababseh D et al (2021) The utility of whole-exome sequencing in accurate diagnosis of neuromuscular disorders in consanguineous families in Jordan. Clin Chim Acta 523:330–338
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25(14):1754–1760
Tarasov A, Vilella AJ, Cuppen E, Nijman IJ, Prins P (2015) Sambamba: fast processing of NGS alignment formats. Bioinformatics 31(12):2032–2034
Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A et al (2013) From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics 43(1110):11.0.1-11.0.33
Wang K, Li M, Hakonarson H (2010) ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38(16):e164
Scott EM, Halees A, Itan Y, Spencer EG, He Y, Azab MA et al (2016) Characterization of Greater Middle Eastern genetic variation for enhanced disease gene discovery. Nat Genet 48(9):1071–1076
Ng PC, Henikoff S (2003) SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res 31(13):3812–3814
Adzhubei I, Jordan DM, Sunyaev SR (2013) Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet 76:7.20.1–7.20.41
Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M (2019) CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res 47(D1):D886–D894
Jaganathan K, KyriazopoulouPanagiotopoulou S, McRae JF, Darbandi SF, Knowles D, Li YI et al (2019) Predicting splicing from primary sequence with deep learning. Cell 176(3):535–48.e24
Plagnol V, Curtis J, Epstein M, Mok KY, Stebbings E, Grigoriadou S et al (2012) A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinformatics 28(21):2747–2754
Godfrey C, Clement E, Mein R, Brockington M, Smith J, Talim B et al (2007) Refining genotype phenotype correlations in muscular dystrophies with defective glycosylation of dystroglycan. Brain 130(Pt 10):2725–2735
Kefi M, Amouri R, Chabrak S, Mechmeche R, Hentati F (2008) Variable cardiac involvement in Tunisian siblings harboring FKRP gene mutations. Neuropediatrics 39(2):113–115
Trovato R, Astrea G, Bartalena L, Ghirri P, Baldacci J, Giampietri M et al (2014) Elevated serum creatine kinase and small cerebellum prompt diagnosis of congenital muscular dystrophy due to FKRP mutations. J Child Neurol 29(3):394–398
Louhichi N, Triki C, Quijano-Roy S, Richard P, Makri S, Meziou M et al (2004) New FKRP mutations causing congenital muscular dystrophy associated with mental retardation and central nervous system abnormalities. Identification of a founder mutation in Tunisian families. Neurogenetics 5(1):27–34
Pegoraro E, Marks H, Garcia CA, Crawford T, Mancias P, Connolly AM et al (1998) Laminin alpha2 muscular dystrophy: genotype/phenotype studies of 22 patients. Neurology 51(1):101–110
Kim MW, Jang DH, Kang J, Lee S, Joo SY, Jang JH et al (2017) Novel mutation (c.8725T>C) in two siblings with late-onset LAMA2-related muscular dystrophy. Ann Lab Med 37(4):359–361
Zambon AA, Ridout D, Main M, Mein R, Phadke R, Muntoni F et al (2020) LAMA2-related muscular dystrophy: Natural history of a large pediatric cohort. Ann Clin Transl Neurol 7(10):1870–1882
Geranmayeh F, Clement E, Feng LH, Sewry C, Pagan J, Mein R et al (2010) Genotype-phenotype correlation in a large population of muscular dystrophy patients with LAMA2 mutations. Neuromuscul Disord 20(4):241–250
Carboni N, Marrosu G, Porcu M, Mateddu A, Solla E, Cocco E et al (2011) Dilated cardiomyopathy with conduction defects in a patient with partial merosin deficiency due to mutations in the laminin-alpha2-chain gene: a chance association or a novel phenotype? Muscle Nerve 44(5):826–828
Fishilevich S, Nudel R, Rappaport N, Hadar R, Plaschkes I, Iny Stein T et al (2017) GeneHancer: genome-wide integration of enhancers and target genes in GeneCards. Database 2017:bax028
Krotoski D, Namaste S, Raouf RK, El Nekhely I, Hindi-Alexander M, Engelson G et al (2009) Conference report: Second conference of the Middle East and North Africa newborn screening initiative: Partnerships for sustainable newborn screening infrastructure and research opportunities. Genetics Med 11(9):663–668
Shawky RM, Elsayed NS, Ibrahim DS, Seifeldin NS (2012) Profile of genetic disorders prevalent in northeast region of Cairo, Egypt. Egypt J Med Hum Genet 13(1):45–62
Temtamy SA, Aglan MS, Meguid NA (2010) Genetic Disorders in Egypt. In: Teebi A (ed) Genetic diseases among Arab populations. Springer, Berlin, pp 219–272
Abdalla EM, Rohrbach M, Bürer C, Kraenzlin M, El-Tayeby H, Elbelbesy MF et al (2015) Kyphoscoliotic type of Ehlers-Danlos Syndrome (EDS VIA) in six Egyptian patients presenting with a homogeneous clinical phenotype. Eur J Pediatr 174(1):105–112
Di Blasi C, Bellafiore E, Salih MA, Manzini MC, Moore SA, Seidahmed MZ et al (2011) Variable disease severity in Saudi Arabian and Sudanese families with c.3924 + 2 T > C mutation of LAMA2. BMC Res Notes 4:534
Allamand V, Sunada Y, Salih MA, Straub V, Ozo CO, Al-Turaiki MH et al (1997) Mild congenital muscular dystrophy in two patients with an internally deleted laminin alpha2-chain. Hum Mol Genet 6(5):747–752
Salih MA (2023) The Meryon Lecture at the 24th annual meeting of the Meryon Society, St. Anne’s College, Oxford, UK, 15th July 2022: Neuromuscular diseases in the Arab population. Neuromuscul Disord 33(10):792–799
Yang H, Kobayashi K, Wang S, Jiao H, Xiao J, Toda T et al (2015) Founder mutation causes classical Fukuyama congenital muscular dystrophy (FCMD) in Chinese patients. Brain Dev 37(9):880–886
Paul L, Rupprich K, Della Marina A, Stein A, Elgizouli M, Kaiser FJ et al (2020) Further evidence for POMK as candidate gene for WWS with meningoencephalocele. Orphanet J Rare Dis 15(1):242
Di Costanzo S, Balasubramanian A, Pond HL, Rozkalne A, Pantaleoni C, Saredi S et al (2014) POMK mutations disrupt muscle development leading to a spectrum of neuromuscular presentations. Hum Mol Genet 23(21):5781–5792
D’Haenens E, Vergult S, Menten B, Dheedene A, Kooy RF, Callewaert B (2022) Expanding the Phenotype of B3GALNT2-Related Disorders. Genes 13(4):694
Kenny TC, Khan A, Son Y, Yue L, Heissel S, Sharma A et al (2023) Integrative genetic analysis identifies FLVCR1 as a plasma-membrane choline transporter in mammals. Cell Metab 35(6):1057–1071
Rajadhyaksha AM, Elemento O, Puffenberger EG, Schierberl KC, Xiang JZ, Putorti ML et al (2010) Mutations in FLVCR1 cause posterior column ataxia and retinitis pigmentosa. Am J Hum Genet 87(5):643–654
De Luca C, Crow YJ, Rodero M, Rice GI, Ahmed M, Lammens M et al (2020) Expanding the clinical spectrum of Fowler syndrome: Three siblings with survival into adulthood and systematic review of the literature. Clin Genet 98(5):423–432
Rullinkov G, Tamme R, Sarapuu A, Laurén J, Sepp M, Palm K et al (2009) Neuralized-2: expression in human and rodents and interaction with Delta-like ligands. Biochem Biophys Res Commun 389(3):420–425
Vargas-Franco D, Kalra R, Draper I, Pacak CA, Asakura A, Kang PB (2022) The Notch signaling pathway in skeletal muscle health and disease. Muscle Nerve 66(5):530–544
Vieira NM, Elvers I, Alexander MS, Moreira YB, Eran A, Gomes JP et al (2015) Jagged 1 Rescues the Duchenne Muscular Dystrophy Phenotype. Cell 163(5):1204–1213
Koutelou E, Sato S, Tomomori-Sato C, Florens L, Swanson SK, Washburn MP et al (2008) Neuralized-like 1 (Neurl1) targeted to the plasma membrane by N-myristoylation regulates the Notch ligand Jagged1. J Biol Chem 283(7):3846–3853
van Reeuwijk J, Grewal PK, Salih MA, Beltrán-Valero de Bernabé D, McLaughlan JM, Michielse CB et al (2007) Intragenic deletion in the LARGE gene causes Walker-Warburg syndrome. Hum Genet 121(6):685–690
Fu X, Yang H, Jiao H, Wang S, Liu A, Li X et al (2017) Novel copy number variation of POMGNT1 associated with muscle-eye-brain disease detected by next-generation sequencing. Sci Rep 7(1):7056
Cauley ES, Pittman A, Mummidivarpu S, Karimiani EG, Martinez S, Moroni I et al (2020) Novel mutation identification and copy number variant detection via exome sequencing in congenital muscular dystrophy. Mol Genet Genomic Med 8(11):e1387
Xiong H, Tan D, Wang S, Song S, Yang H, Gao K et al (2015) Genotype/phenotype analysis in Chinese laminin-α2 deficient congenital muscular dystrophy patients. Clin Genet 87(3):233–243
Acknowledgements
We thank the families for their participation in our study. The research was supported by funding from the National Institutes of Health (grant #R01NS109149) and the Robert Wood Johnson Foundation (grant #74260) to M.C.M., and by a fellowship from the Egyptian Ministry of Higher Education to S.S.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Safwat, S., Flannery, K.P., El Beheiry, A.A. et al. Genetic blueprint of congenital muscular dystrophies with brain malformations in Egypt: A report of 11 families. Neurogenetics (2024). https://doi.org/10.1007/s10048-024-00745-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10048-024-00745-z