
Abstract
Tiller number greatly contributes to grain yield in wheat. Using ethylmethanesulfonate mutagenesis, we previously discovered the oligo-tillering mutant ot1. The tiller number was significantly lower in ot1 than in the corresponding wild type from the early tillering stage until the heading stage. Compared to the wild type, the thousand-grain weight and grain length were increased by 15.41% and 31.44%, respectively, whereas the plant height and spike length were decreased by 26.13% and 37.25%, respectively. Transcriptomic analysis was conducted at the regreening and jointing stages to identify differential expressed genes (DEGs). Functional enrichment analysis with the Kyoto Encyclopedia of Genes and Genomes (KEGG) and Gene Ontology (GO) databases showed differential expression of genes associated with ADP binding, transmembrane transport, and transcriptional regulation during tiller development. Differences in tiller number in ot1 led to the upregulation of genes in the strigolactone (SL) and abscisic acid (ABA) pathways. Specifically, the SL biosynthesis genes DWARF (D27), D17, D10, and MORE AXILLARY GROWTH 1 (MAX1) were upregulated by 3.37- to 8.23-fold; the SL signal transduction genes D14 and D53 were upregulated by 1.81- and 1.32-fold, respectively; the ABA biosynthesis genes 9-CIS-EPOXICAROTENOID DIOXIGENASE 3 (NCED3) and NCED5 were upregulated by 1.66- and 3.4-fold, respectively; and SNF1-REGULATED PROTEIN KINASE2 (SnRK2) and PROTEIN PHOSPHATASE 2C (PP2C) genes were upregulated by 1.30- to 4.79-fold. This suggested that the tiller number reduction in ot1 was due to alterations in plant hormone pathways. Genes known to promote tillering growth were upregulated, whereas those known to inhibit tillering growth were downregulated. For example, PIN-FORMED 9 (PIN9), which promotes tiller development, was upregulated by 8.23-fold in ot1; Ideal Plant Architecture 1 (IPA1), which inhibits tiller development, was downregulated by 1.74-fold. There were no significant differences in the expression levels of TILLER NUMBER 1 (TN1) or TEOSINTE BRANCHED 1 (TB1), indicating that the tiller reduction in ot1 was not controlled by known genes. Our findings provide valuable data for subsequent research into the genetic bases and regulatory mechanisms of wheat tillering.
Similar content being viewed by others

Introduction
Wheat (Triticum asetivum L.) is one of the most important staple crops throughout the world, providing ~ 20% of all food calories consumed by humans (Shiferaw et al. 2013). Wheat yield is determined by the thousand-grain weight, the number of spikes per unit, and the number of grains per spike (Shang et al. 2021). The number of spikes per unit is positively correlated with tiller number. Wheat tillers are lateral branches, that grow from the main stem (Luo et al. 2021). Despite the association between tiller number and yield, lower-tillering mutants can also have high yield. The thousand-grain weights of lines with fewer tillers are increased by an average of 57.43% (Richards 1988), and the yields of oligo-tillering lines are increased by 11.90% compared to multi-tillering lines under drought stress conditions (Mitchell et al. 2013). The tiller inhibition 4 (tin4) mutant exhibits oligo-tillering, but its maximum yield is 2.26–13.33% higher than the yield of multi-tillering lines under high-density planting conditions (Wang et al. 2022). A deeper understanding of the genetic mechanisms underlying wheat tillering could contribute to the genetic improvement of wheat yield.
The genes that control tillering in rice are better understood than those in wheat. MONOCULM 1 (MOC1) and MONOCULM 3 (MOC3) regulate the initiation and outgrowth of axillary buds (Li et al. 2003; Shao et al. 2019). FLORAL ORGAN NUMBER1 (FON1) also regulates axillary bud outgrowth, which promotes tiller growth. MOC1 and MOC3 co-activation induces FON1 expression (Moon et al. 2006; Shao et al. 2019). LAX PANICLE 1 (LAX1) and LAX PANICLE 2 (LAX2) regulate tiller number by promoting differentiation of axillary meristem cells (Oikawa and Kyozuka 2009; Tabuchi et al. 2011). TEOSINTE BRANCHED 1 (OsTB1) encodes the TCP transcription factor, which is primarily expressed in axillary buds. OsTB1 is a negative regulator of rice lateral branching (Takeda et al. 2003). OsTB1 directly inhibits OsD14 expression and regulates tiller growth by interacting with the MADS-box domain of MADS57 (Guo et al. 2013). Overexpression of HEME ACTIVATOR PROTEIN (OsHAP2E) increases the photosynthesis rate and tiller number (Alam et al. 2015). PALE GREEN LEAF (PGL)/CAO1 encodes chlorophyllide a oxygenase; pgl mutants show inhibited axillary bud growth and decreased tiller number (Yang et al. 2016). OsbHLH025/DPF encodes a basic helix–loop–helix (bHLH) transcription factor and promotes diterpenoid phytoalexins production. DPF overexpression is associated with decreased tiller number (Yamamura et al. 2015). PIN-FORMED (OsPIN2), OsPIN5b, and OsPIN9 regulate auxin transporters; PIN2 and PIN9 overexpression increases tiller number (Chen et al. 2012; Hou et al. 2021), whereas PIN5b overexpression decreases tiller number (Lu et al. 2015).
Research into wheat tillering has advanced in recent years. Several genes controlling oligo-tillering have been identified in mutants, such as TILLER NUMBER 1 (tn1) (Dong et al. 2023), tin1 (Richards 1988), tin2 (Peng et al. 1998), tin3 (Kuraparthy et al. 2006; Ahmed et al. 2023), tin4 (Wang et al. 2022), tin5 (Si et al. 2022), tin6 (Schoen et al. 2023), fertile tiller inhibition gene (ftin) (Zhang et al. 2013), and dwarf-monoculm (dmc) (An et al. 2019). Morphological studies have revealed that the decreased tiller numbers observed in tin1, tin4, tin5, ftin, and Low number of tillers 1 (lnt1) (Dabbert et al. 2010) mutants are due to inhibited and abnormal axillary bud development. In tin1, tiller outgrowth ceases when the shoot apex transitions from the vegetative to the reproductive stage (Kebrom et al. 2012; Richards 1988). ftin shows a normal tiller number at the seedling stage, but very few tillers are observed at heading stage (Zhang et al. 2013). This suggests that a lower effective tiller number is not due to axillary bud differentiation, but to delayed tiller outgrowth and development at later stages. In tin4, tin5, and lnt1, decreases in tiller number are due to inhibition of secondary tiller bud development (Dabbert et al. 2010; Si et al. 2022; Wang et al. 2022). In contrast, both tiller bud differentiation and development are inhibited in dmc (An et al. 2019). In tin6, tiller number is decreased at the seedling and heading stages (Schoen et al. 2023). Genes that regulate wheat tillers have pleiotropic effects for plant height and other yield related traits. For example, significantly higher thousand-grain weight, grain length, and grain width have been identified in lower-tillering wheat mutants (Kuraparthy et al. 2006; Wang et al. 2022).
Strigolactones (SLs) are phytohormones produced from plant carotenoid derivatives (Al-Babili and Bouwmeester 2015). SLs and their precursors affect many aspects of plant architecture and development, including tiller formation and development and shoot branching. A lack of SLs can increase tiller and shoot branching numbers, whereas excessive SL levels can inhibit tiller growth (Gomez-Roldan et al. 2008; Zhao et al. 2019). Excessive SL levels also promote lateral root formation and hair elongation (Kapulnik et al. 2011; Sun et al. 2016). Furthermore, SLs play important roles in rhizosphere signaling through effects such as inducing hyphal branching among arbuscular mycorrhizal fungi and stimulating germination of the parasitic plants Striga spp. and Orobanche spp. (Fiorilli et al. 2019; Zhao et al. 2019). Several genes in SL biosynthesis and signal transduction pathways have been identified in rice and Arabidopsis thaliana. SL biosynthesis is regulated by four main enzymes. The first step is reversible isomerization of all-trans-into 9-cis-β-carotene, which is catalyzed by DWARF27 (D27) (Abuauf et al. 2018). The product undergoes cleavage and rearrangement by CAROTENOID CLEAVAGE DIOXYGENASE 7 (CCD7)/D17 and CAROTENOID CLEAVAGE DIOXYGENASE 8 (CCD8)/D10, respectively, yielding carlactone (Alder et al. 2012). Carlactone is catalyzed of cytochrome P450 monooxygenase, after which MORE AXILLARY GROWTH 1 (MAX1) and other enzymes act sequentially to form canonical and non-canonical SLs such as carlactonoic acid, 4-deoxyorobanchol, and Orobanchol (Booker et al. 2005). D3, D14, and D53 are involved in SL signal transduction (Ishikawa et al. 2005; Jiang et al. 2013; Zhao et al. 2013), which is initiated by the α/β-sheet hydrolase receptor portion of protein D14. D14 binds D3 through the C-terminal helix structure (Shabek et al. 2018), then combines with D53 to form a complex for SL signal transduction (Tal et al. 2022; Zhou et al. 2013). CYTOKININ OXIDASE/DEHYDROGENASE 9 (CKX9) and Ideal Plant Architecture 1 (IPA1) are the downstream genes that directly participate in SL signal transduction (Duan et al. 2019; Song et al. 2017).
Abscisic acid (ABA) is derived from β-carotene through a series of enzymatic reactions. This process begins with hydroxylation of all-trans-β-carotene and results in zeaxanthin formation. Subsequently, ABA DEFICIENT 1 (ABA1) converts all-trans-zeaxanthin to all-trans-violaxanthin (Barrero et al. 2005). ABA DEFICIENT 4 (ABA4) is involved in the conversion of all-trans-violaxanthin to all-trans-neoxanthin (Dall’Osto et al. 2007), and an ABA4 homolog affects branching and adventitious root formation (Ma et al. 2014). Carotenoids with the all-trans configuration are then isomerized to the 9-cis counterparts, which are catalyzed by 9-CIS-EPOXICAROTENOID DIOXIGENASE 3 (NCED3) and 9-CIS-EPOXICAROTENOID DIOXIGENASE 5 (NCED5) to form xanthoxin (Bang et al. 2013; Zhu et al. 2009). Xanthoxin is further oxidized by ABA DEFICIENT 2 (ABA2) to produce abscisic aldehyde; this is in turn oxidized by abscisic aldehyde oxidases (AAOs) and ABA DEFICIENT 3 (ABA3) to produce ABA (Nambara et al. 1998; Watanabe et al. 2018). PYR-LIKE (PYLs) and PROTEIN PHOSPHATASE 2C (PP2Cs) also play crucial roles in ABA perception and signal transduction (Bhatnagar et al. 2017; Kim et al. 2012). ABA forms complexes with PYLs and PP2Cs, enabling the release of PP2C-mediated SNF1-REGULATED PROTEIN KINASE 2 (SnRK2) inhibition. PYLs, PP2Cs, and SnRK2s are the three core protein elements in the ABA signaling pathway (Chen et al. 2020).
RNA-sequencing (RNA-seq) has been widely used to identify differentially expressed genes (DEGs) between biological samples. DEGs can reveal the genetic mechanisms underlying phenotypic changes. In the present study, an oligo-tillering mutant, ot1, was developed in the winter wheat cultivar “Jing411” via ethyl methane sulfonate (EMS) treatment. Compared with the corresponding wild type (WT), ot1 exhibited decreased tiller number and plant height but increased thousand-grain weight. The tiller bud tissues of ot1 and WT plants were sampled and characterized at the transcriptomic level via RNA-seq. This analysis suggested that the tiller number inhibition in ot1 may have been correlated with the SL and ABA pathways. The results of this study could guide future research into the regulatory mechanisms controlling wheat tillering.
Materials and methods
Plant materials
The oligo-tillering mutant ot1 was discovered in a previously generated EMS population (Guo et al. 2017) using the winter wheat cultivar “Jing411” as the parent. ot1 was purified through multiple generations of self-pollination; the stable M7 line was used in transcriptomic and pathway regulation analyses. This mutant was also crossed with the WT, to develop the multi-tiller and oligo-tiller BC2F2 and BC2F3 lines, which were used to verify gene expression patterns. The mutant, WT, and BC2F3 lines were planted at the experimental station of the Institute of Crop Sciences, Chinese Academy of Agricultural Sciences (Beijing, China). Ten rows of the mutant and WT were planted and two rows of each BC2F3 line were planted. Each row was 2 m length and there was 10 cm between individual plants.
Investigation of tillering and other agronomic traits
Tiller number was counted in 10 ot1 and 10 WT plants from the regreening stage until the heading stage. After harvesting, 10 individuals of each genotype were selected for measurements of plant height, spike length, thousand-grain weight, grain width, and grain length. Tiller number was counted in the BC2F3 lines at 66 d after sowing for 10 individuals from each line. These values were compared with those of the corresponding parents.
Statistical analyses
All statistical analyses were conducted in GraphPad Prism 8 v8.0.2. Differences between pairs of groups were assessed with Student’s t test and considered significant at p < 0.05.
Sampling for RNA-seq
Axillary buds were collected from ot1 and WT plants at the regreening stage (141 d after sowing) and the jointing stage (170 d after sowing). The tiller buds of four individual plants were collected and mixed to form a single sample; there were three replicates per genotype at each stage for a total of 12 samples. These were used for RNA-seq and validation with real-time quantitative PCR (RT-qPCR). RNA was extracted using the RNeasy Plant Mini Kit (Qiagen, Germany). RNA quantity and integrity were measured with a Nanodrop 2000 (Thermo Fisher Scientific, Waltham, MA, USA) and a Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA, USA), respectively.
The tiller buds of multi-tiller and oligo-tiller BC2F3 lines and their parents were also collected at the seedling stage (66 d after sowing) for RT-qPCR verification. For this, three BC2F3 lines were combined to form a single sample.
RNA-seq and related analyses
RNA-seq libraries were constructed using the Illumina RNA Library Prep Kit (Vazyme, Nanjing, China) following the manufacturer’s instructions. Poly(A)-containing transcripts were enriched from the total RNA with Oligo(dT)-coated magnetic beads. For each sample, adapters with unique barcodes were ligated to the end-polished cDNA fragments. The libraries were amplified with PCR and quantitated on a Qubit 2.0 (Thermo Fisher Scientific, Waltham, MA, USA). RNA libraries were sequenced on the Illumina NovaSeq platform, producing raw reads in FASTQ format. Sequencing was conducted by Tcuni (Chengdu, China). Adapters and low-quality data were removed using fastp (https://github.com/OpenGene/fastp) with default parameters. The Q30 scores and GC contents of the clean data were calculated simultaneously. Principal component analysis (PCA) was conducted on the clean data using BWKCloud tools (https://www.biocloud.net/fxpt/app).
Quantitative sample analysis was performed with kallisto (https://pachterlab.github.io/kallisto/) using the default parameters (Bray et al. 2016) to obtain gene expression values. Differences between sample types were then analyzed with the R package “edgeR3” (https://bioconductor.org/packages/release/bioc/html/edgeR.html) using thresholds of false discovery rate (FDR) ≤ 0.05 and |Log2(fold change [FC])| ≥ 1 (Robinson et al. 2010). The resulting DEGs were used in subsequent functional analyses.
Gene Ontology (GO) term enrichment analyses were conducted with the R package “Goseq” (Young et al. 2010), and Kyoto Encyclopedia of Genes and Genomes (KEGG) biochemical pathway enrichment analyses were carried out with KOBAS (Xie et al. 2011). GO and KEGG terms enriched at a corrected p value < 0.05 were considered statistically significant.
RT-qPCR
Samples used in RNA-seq were validated with RT-qPCR. After genomic DNA was removed from the RNA samples, first-strand cDNA was synthesized using the All-in-One First-Strand cDNA Synthesis SuperMix Kit (TransGen Biotech, China). RT-qPCR was then conducted with the PerfectStart Green qPCR SuperMix Kit (TransGen Biotech, China) on the CFX 96 Real-Time System (Bio Rad, Hercules, CA, USA) with following thermocycling program: one cycle of 30 s at 94 ℃ followed by 45 cycles of 5 s at 94 ℃, 15 s at 60 ℃, and 10 s at 72 ℃. A melting curve analysis was conducted at the end of the 45 cycles. Reverse transcription and qPCR procedures were performed following the manufacturers’ instructions. RT-qPCR was carried out in technical triplicate. Relative expression levels were calculated using the 2−ΔΔCt method (Livak and Schmittgen 2001) with normalization to the internal control gene TaActin. All primers used for RT-qPCR are shown in Table S1.
Expression correlation coefficient analysis
Correlations of expression levels of DEGs between the RNA-seq and RT-qPCR data were calculated. The fold change of DEGs in the mutant and WT from RNA-seq or RT-qPCR was calculated by gene expression levels in the mutant divided by that in the WT. The correlations of RNA-seq and RT-qPCR data were analyzed using GraphPad Prism 8 v8.0.2. Correlations of gene expression between BC2F3 lines and the corresponding parent lines from the RT-qPCR data were also calculated based on the gene expression levels from oligo-tiller lines divided by that from multi-tiller lines.
Results
ot1 had a significantly lower-tiller number but increased thousand-grain weight compared to the WT
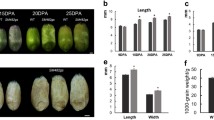
The mutant line ot1 exhibited fewer tillers than the corresponding WT (Fig. 1a, b). From the seedling until the heading stage, the tiller number did not significantly increase in ot1 as the plants grew (Fig. 1c). Furthermore, the ot1 spike number was only 9.31% of the spike number in the WT at the heading stage (Fig. 1d).

Phenotypic variations in ot1. a ot1 and wild-type (WT) at the seedling stage. Scale bar = 5 cm. b ot1 and WT plants at the heading stage. Scale bar = 20 cm. c Dynamic changes in tiller number from the regreening stage to the heading stage. d Effective tiller number at the heading stage. e Plant height. f Spike length. g Grain length and width. Scale bar = 1 cm. h Thousand-grain weight. i Grain length. j Grain width. Data are presented as the mean ± standard deviation (SD). *p < 0.05, **p < 0.01; ns, no significant difference (two-tailed Student’s t-test)
ot1 also showed a significantly decreased plant height and spike length compared to the WT (Fig. 1e, f). Specifically, the average plant height was 66.86 cm, representing a 26.13% decrease compared to the WT, and the ot1 spike length was decreased by 37.25%. However, the thousand-grain weight and grain length were increased by 15.41% and 31.44%, respectively, compared to the WT (Fig. 1g–i). No significant variation was detected in grain width (Fig. 1j). This indicated that grain length but not width contributed to the increased thousand-grain weight of ot1 mutants.
RNA-seq data were of high quality
A total of 162.60 Gb of clean data were obtained, ranging from 12.56 to 15.05 Gb per sample. The GC content of the 12 samples ranged from 52.50 to 53.20% and the average Q30 coverage was 92.33% (Table S2). These results indicated that the RNA-Seq data were suitable for further analyses.
DEGs between ot1 and WT
At the regreening stage, 5582 DEGs were identified in ot1 compared to WT plants. Of these, 4865 were upregulated and 717 were downregulated (Table S3). TraesCS6B03G1086300LC was the most strongly downregulated gene in the ot1 with a Log2(FC) value of 8.74. This gene encodes a protein of unknown function. The most strongly upregulated gene in ot1 was TraesCSU03G0251900, which encodes a protein modifier of SNC11-like.
At the jointing stage, there were 3319 DEGs in ot1 compared to WT plants (Fig. 2a, b). Among them, 2457 were upregulated and 862 were downregulated (Table S4). TraesCS6A03G1022000LC was the most strongly downregulated in ot1; this gene encodes a protein of unknown function. TraesCS2B03G0186800LC, which also encodes a protein of unknown function, was the most strongly upregulated gene in ot1.

Transcriptomic analysis at the regreening and jointing stages. a Relative expression of differentially expressed genes (DEGs) at the regreening stage. b Relative expression of DEGs at the jointing stage. c Correlation analysis of expression levels between ot1 and WT plants. d Principal component analysis (PCA) of ot1 and WT plants at each sampled stage. GS, regreening stage; JS, jointing stage
Cluster analysis indicated that the gene expression patterns among biological replicates were similar (Fig. 2c), demonstrating the replicability and reliability of the transcriptomic data. PCA also showed significant differences between the four groups. The first and second principal components (PC1 and PC2, respectively) accounted for 30.56% and 25.37% of the variance, respectively (Fig. 2d).
Genes involved in ADP binding, transmembrane transport, and transcriptional regulation were differentially expressed in ot1
To further explore biological pathways or processes related to wheat tillering, GO enrichment analysis was performed on the DEGs at both stages. GO terms were assessed in three categories: biological process (BP), molecular function (MF), and cellular component (CC) terms.
At the regreening stage, the upregulated DEGs were classified into 377 BP terms, 354 MF terms, and 72 CC terms (Table S5). The most significantly enriched were the BP terms, which included metal ion transport, zinc ion transmembrane transport, and transmembrane transport (Fig. 3a). Among the downregulated DEGs, 121 BP terms, 141 MF terms, and 44 CC terms were significantly enriched (Table S6). Here again, the BP terms were most significantly enriched and included the terms “metabolic process,” “cell wall macromolecule catabolic process,” and “chitin catabolic process.” Genes related to ADP binding and transmembrane transport were also significantly enriched, as were genes associated with ion transmembrane transport, the cell membrane, and cell wall composition (Fig. 3b).

Significantly enriched Gene Ontology (GO) terms in DEGs between ot1 and WT plants at the regreening and jointing stages. a, b Significantly enriched biological process (BP), molecular function (MF), and cellular component (CC) terms in the a upregulated and b downregulated DEGs at the regreening stage. c, d Significantly enriched BP, MF, and CC terms in the c upregulated and d downregulated DEGs at the jointing stage. GS, regreening stage; JS, jointing stage
At the jointing stage, the upregulated DEGs were significantly enriched in BP terms, including regulation of systemic acquired resistance, transcription, and the glyoxylate cycle (Fig. 3c). The most highly enriched BP terms in the downregulated DEGs were cell redox homeostasis, regulation of transcription, and response to biotic stimulus (Fig. 3d). Genes associated with ADP binding, transcription regulation, and DNA binding were significantly upregulated in the axillary buds. This suggested that tiller bud growth was regulated by various transcription factors at this stage. HAP2E and MOC1 are transcription factors associated with transcriptional regulation and DNA binding; these genes are known to control tiller growth (Li et al. 2003; Alam et al. 2015). Here, the homeologs HAP2E-A and HAP2E-B were upregulated in ot1 mutants by 1.52-fold and 1.58-fold, respectively, and MOC1-D was upregulated by 1.12-fold compared to the WT.
ot1 mutation altered metabolic and phytohormone signal transduction pathways
KEGG biochemical pathway enrichment analyses were next performed in the DEGs at both stages to explore biochemical pathways involved in tiller growth. At the regreening stage, the 5582 DEGs were involved in 117 pathways (Table S7). The 10 most highly enriched pathways were metabolic pathways (taes01100), biosynthesis of secondary metabolites (taes01110), glutathione metabolism (taes00480), phenylpropanoid biosynthesis (taes00940), linoleic acid metabolism (taes00591), the MAPK signaling pathway (taes04016), nitrogen metabolism (taes00910), alpha-Linolenic acid metabolism (taes00592), cyanoamino acid metabolism (taes00460), and arginine and proline metabolism (taes00330) (Fig. 4a). At the jointing stage, the 3319 DEGs were annotated as involved in 109 pathways (Table S8). The 10 most strongly enriched pathways were plant hormone signal transduction (taes04075), phenylalanine metabolism (taes00360), biosynthesis of secondary metabolites (taes01110), glyoxylate and dicarboxylate metabolism (taes00630), phenylpropanoid biosynthesis, alanine (taes00940), aspartate and glutamate metabolism (taes00250), plant-pathogen interaction (taes04626), glutathione metabolism (taes04626), isoquinoline alkaloid biosynthesis (taes00480), and stilbenoid, diarylheptanoid, and gingerol biosynthesis (taes00950) (Fig. 4b).

Significantly enriched Kyoto Encyclopedia of Genes and Genomes (KEGG) biochemical pathways among DEGs between ot1 and WT plants at the regreening stage and jointing stages. a Enriched KEGG pathways among DEGs at the regreening stage. b Enriched KEGG pathways among DEGs at the regreening stage. GS, regreening stage; JS, jointing stage
Differential expression of genes related to phytohormone synthesis and signal transduction in ot1
DEGs related to the SL, ABA, indole-3-acetic acid (IAA), ethylene (ETH), and jasmonate (JA) biosynthesis and signal transduction pathways were identified. The expression levels of selected DEGs were validated by RT-qPCR. Correlation analysis of expression level of each gene from RNA-seq and RT-qPCR data revealed that a high correlation (R2 = 0.85) was observed between these two data (Fig S1a, b).
Transcriptomic analysis revealed significant upregulation of the carotenoid oxygenase gene in ot1 (Table S9). Because both SL and ABA are generated from carotenoid derivatives (Barrero et al. 2005; Yoneyama et al. 2018), we further investigated the expression levels of genes involved in the ABA and SL biosynthesis and signaling pathways. For these pathways, expression levels of some genes were also validated with RT-qPCR (Fig. 5, Table S10). Compared with the WT, the SL biosynthesis genes TaD27-A, TaD27-B, and TaD27-D were significantly upregulated (by 1.96- to 3.37-fold) in ot1; TaD17-A, TaD17-B, and TaD17-D were upregulated by 1.79- to 4.27-fold, and TaD10-A, TaD10-B, and TaD10-D were upregulated by 5.71-, 2.67-, and 5.34-fold, respectively (Fig. 5c–f). Furthermore, TaMAX1-A, TaMAX1-B, and TaMAX1-D were significantly upregulated by 3.94- to 8.23-fold in ot1. This suggested that genes involved in SL biosynthesis were expressed at significantly higher levels in ot1 than WT plants. The SL signal transduction TaD14 and TaD53 genes were also significantly upregulated. Specifically, TaD14-A, TaD14-B, TaD14-D, and TaD53-A were significantly upregulated by 1.44-, 1.81-, 1.23-, and 1.32-fold, respectively (Fig. 5g–i). CKX9 is a downstream gene of D53; the wheat subgenomes A, B, and D contain three homeologs of CKX9 namely TaCKX9-A, TaCKX9-B, and TaCKX9-D, respectively. These three genes were significantly upregulated by 2.07-, 2.16-, and 1.95-fold, respectively (Fig. 5j).

Expression levels of genes related to strigolactone (SL) biosynthesis and signal transduction as determined with real-time quantitative PCR (RT-qPCR). a Expression levels of genes involved in SL pathways at the regreening and jointing stages. b Model of the SL biosynthesis and signaling pathways. c–j Expression levels of c TaD27-7A, d TaD17-2B, e TaD17-2B, f TaD10-3A, g TaD14-4A, h TaD14-4B, i TaD53-A, TaD53-B, and TaD53-D, and j TaCKX9-A, TaCKX9-B, and TaCKX9-D. GS, regreening stage; JS, jointing stage. Color indicates expression values in Log2(TPM). Data are presented as the mean ± SD, *p < 0.05, **p < 0.01 (two-tailed Student’s t-test)
These results suggested that the expression levels of genes associated with the SL pathway were significantly altered in ot1 mutants, which had abnormal tiller development. Furthermore, compared with the regreening stage, TaMAX1, TaD14, and TaCKX9 were upregulated by 37.27%, 37.40%, and 122.23%, respectively at the jointing stage.
It has previously been demonstrated that ABA can inhibit the growth and development of lateral buds. The ABA biosynthesis genes ABA1, ABA2, ABA3, and ABA4 were significantly upregulated in ot1 compared to the WT (Fig. 6b–e). NCED3 and NCED5 are reportedly involved in the ABA biosynthesis pathway (Dong et al. 2023; Zhu et al. 2009); these two genes were upregulated by 1.66- and 3.4-fold, respectively, in ot1 compared to the WT (Fig. 6a, i). Furthermore, in ot1, the average NCED3 and NCED5 expression levels at the jointing stage were 4.81- and 3.23-fold higher, respectively, than in the regreening stage. Genes involved in ABA signal transduction, such as PP2Cs and SnRK2s, were also significantly upregulated in ot1 compared to the WT (Fig. 6f, g). For example, PP2Cs were upregulated by 1.9- to 4.79-fold, and SnRK2s were upregulated by 2.09- to 2.79-fold. In contrast, PYL was significantly downregulated by 70% (Fig. 6h, Table S11). These differences in gene expression between genotypes suggested that the ABA biosynthesis and signaling pathways may have been enhanced in ot1. There were no significant differences in PYL, PP2C, or SnRK2 expression levels in ot1 between the regreening stage and the jointing stage.

Expression levels of DEGs associated with abscisic acid (ABA) biosynthesis and signal transduction and as determined with RT-qPCR. a Expression levels of genes involved in the ABA pathway at the regreening and jointing stages. b–i Expression levels of b ABA1, c ABA2, d ABA3, e ABA4, f PP2Cs, g PP2Cs5, h PYL, and i TaNCED3. GS, regreening stage; JS, jointing stage. Color indicates expression values in Log2(TPM). Data are presented as the mean ± SD. *p < 0.05, **p < 0.01 (two-tailed Student’s t-test)
There was a total of 111 DEGs related to auxin biosynthesis and signaling in ot1 (Table S12). These included 13 IAA biosynthesis genes; of seven such genes encoding flavin monooxygenase-like proteins, four were upregulated and three were downregulated. The other six genes, which encoded aldehyde oxidase/xanthine dehydrogenase proteins, were all upregulated. The remaining 98 genes were involved in auxin signaling and comprised 11 PINs involved in IAA transport, 21 genes encoding AUX/IAA proteins as auxin response factors, 51 genes in the auxin-responsive SAUR gene family, nine genes encoding GH3 family proteins, and six genes encoding auxin-repressed proteins.
DEGs in the ETH and JA pathways were also investigated. Two genes encoding ethylene-responsive binding factors and four genes encoding ethylene insensitive protein were significantly upregulated in ot1 (Table S13). Additionally, 36 genes encoding proteins with the Tify domain that were involved in the JA pathway were significantly upregulated in ot1 (Table S14). Such differences in the expression levels of genes associated with hormone synthesis and signaling pathways could partially explain the oligo-tillering phenotype of ot1.
Expression levels of known tillering development regulatory genes were not consistent with tiller variations
We also examined the expression levels of other genes known to be involved in tiller development (Table S15, Fig. 7a–d). Intriguingly, compared with WT, expression levels of genes known to promote tiller development were significantly upregulated in the mutant, whereas genes known to inhibit tiller development were significantly downregulated. For example, HAP2E, PIN2, PIN9, CAO1, MOC1, and FON1, which are known to promote tillering in rice, were all significantly upregulated (from 1.11- to 8.23-fold) in ot1 (Fig. 7a, b). The genes IPA1, PIN5b, and bHLH025 are known to inhibit tillering in rice; here, IPA1 and bHLH025 were significantly downregulated in ot1, although PIN5b was significantly upregulated (by 3.70-fold). There were no significant differences in TN1 or TB1 expression (Fig. 7c, d).

Expression levels of DEGs involved in tiller development as determined with RT-qPCR. a Expression levels of genes involved in the ABA pathway at the regreening and jointing stages. b–d Expression levels of b TaPIN9, c TN1, and d TB1. GS, regreening stage; JS, jointing stage. Color indicates expression values in Log2(TPM). Data are presented as the mean ± SD. *p < 0.05, **p < 0.01 (two-tailed Student’s t-test)
Although there were no significant differences in the expression of TB1 between ot1 and WT plants at either stage, expression of this gene did gradually increase along with tiller development. Compared to the regreening stage, TB1 was upregulated by 1.52-fold at the jointing stage. Similarly, PIN2, IPA1, and MOC1 were upregulated by an average of 2.92-, 2.18-, and 3.16-fold, respectively, at the jointing stage.
Gene expression patterns in BC2F3 lines were consistent with those of the corresponding parents
The BC2F3 populations with oligo- and multi-tillers (Fig S2) were further used to validate the relationship between gene expression levels and tillering. Correlation analysis showed that expression levels of key genes were highly correlated between the BC2F3 lines and the corresponding parents (Fig S1c, d). There were no significant differences detected in TN1 or TB1 between the multi- and oligo-tiller lines. Expression levels of eight genes (TaD10-3A, ABA1, PYL, TaCKX9-B, TaCKX9-D, IPA1-A, IPA1-B, and IPA1-D) were consistent between the BC2F3 lines and their parents (Fig. 8a). Expression levels of five genes (TaD17-2D, TaD14-4B, ABA2, TaNCED3, and PP2Cs5) were not consistent with the transcriptomic data, but were consistent with the corresponding parents (Fig. 8b). This suggested that those genes may also have been regulated by the growth period. The expression patterns of TaD27-7A and PP2Cs were inconsistent between the two sampling stages and among genotypes (Fig. 8c), suggesting that they may not have regulated tiller development in ot1.

Expression patterns in BC2F3 lines as determined with RT-qPCR. a Genes with consistent expression patterns between multi-tiller and oligo-tiller lines and across sampling stages. b Genes with consistent expression patterns between multi-tiller and oligo-tiller lines. c Genes with inconsistent expression patterns. Data are presented as the mean ± SD. *p < 0.05, **p < 0.01; ns, no significant difference (two-tailed Student’s t-test)
Discussions
The ot1 phenotype differed from those of known lower-tiller wheat mutants
The mutants dmc and tin6 are characterized by mono-tillering and dwarfism from the seedling stage to the heading stage (An et al. 2019; Schoen et al. 2023). In contrast, the ot1 mutant maintained an average of three tillers. This indicated that it retained the ability to form tillers and that the tiller inhibitory effect in this mutant therefore differed from those in other mutants. In tin4 mutants, the tiller number is significantly decreased at the seedling stage and does not increase at all until the heading stage (Wang et al. 2022). This similar to the variation in tiller number observed in ot1, but plant height and spike length are not changed in tin4. ftin possesses normal tillering ability at the seedling stage but has a reduced number of effective tillers after heading (Zhang et al. 2013), which again differs from ot1. Previous studies have observed that reductions in tiller number result from inhibition of tiller bud differentiation or growth. To understand the mechanisms leading to reduced tiller number in ot1, structural variations in the ot1 tiller buds must be examined from the very beginning of the seedling stage.
Hormone biosynthesis and signal transduction pathways were enhanced in ot1
Phytohormones are some of the important factors that regulate plant growth and development. Tiller bud growth and differentiation are regulated by multiple genes and hormones, including SLs (Al-Babili and Bouwmeester 2015). Canonical SLs are primarily associated with inter-root communication in plants, whereas non-canonical SLs mainly affect tiller differentiation and growth (Ito et al. 2022). Non-canonical SLs are formed by D27, D17, and D10, then are converted to canonical SLs through a multi-step biosynthetic process involving MAX1 (Zhang et al. 2014). ot1 tiller buds showed significant upregulation of TaD27, TaD17, TaD10, and TaMAX1 at the regreening stage, demonstrating enhancement of the SL biosynthesis pathway in ot1. Compared with the regreening stage, TaMAX1 was upregulated at the jointing stage. This indicated that canonical SL biosynthesis was enhanced at the jointing stage. SL signaling relies on a complex formed by D53, D14, and D3 (Shabek et al. 2018). TaD14 and TaD53 were significantly upregulated in ot1 compared to the WT, indicating enhancement of the SL signaling pathway in addition to the biosynthetic pathway. OsCKX9 functions downstream of D53 and is involved in SL signaling. OsCKX9 is significantly upregulated in response to increased endogenous SL content or to treatment with the SL analogue GR24 (Duan et al. 2019). IPA1 also functions downstream of D53 and regulates tillering in rice (Song et al. 2017). D53 and TaCKX9 were significantly upregulated in ot1 compared to WT plants, whereas IPA1 was downregulated. These outcomes were consistent with previous findings in rice, demonstrating the enhanced function of the SL signaling pathway in ot1 tiller buds.
ABA reportedly plays an important role in inhibiting tillering and branch growth (Abuauf et al. 2018; Liu et al. 2020). Increased endogenous ABA contents can reduce tillering in wheat, rice (Liu et al. 2020), and Arabidopsis (Yao and Finlayson 2015). NCED3 is a key gene in the ABA biosynthetic pathway; compared with the WT, the Arabidopsis mutant nced3 has longer axillary buds and more branches (González-Grandío et al. 2017). Here, ABA1, ABA2, ABA3, ABA4, NCED3, and NCED5 were all upregulated in ot1 at both sampling stages compared with the WT. The expression levels of NCED3 and NCED5 were also significantly increased at the jointing compared to the regreening stage. These results demonstrate increased expression of ABA biosynthesis genes in ot1 in addition to gradual increases in ABA synthesis gene expression during tiller development. In wheat, ABA signal transduction is dependent on PYLs, PP2Cs, and SnRK2s (Bhatnagar et al. 2017; Kim et al. 2012; Chen et al. 2020). PP2Cs and SnRK2s were here found to be significantly upregulated in ot1, suggesting enhancement of the ABA signaling pathway. However, there were no differences in the expression of ABA signaling genes between the two sampled stages in ot1.
IAA accumulation inhibits tiller bud growth. The IAA efflux genes PIN2, PIN9, and PIN5b were differentially expressed in ot1 compared to the WT. PIN2 and PIN9 regulate IAA transportation out of the tiller bud and promote tiller bud growth (Chen et al. 2012; Hou et al. 2021). PIN2 and PIN9 were upregulated in ot1 mutant, suggesting that these genes did not negatively affect ot1 tiller growth. In contrast to PIN2 and PIN9, PIN5b negatively regulates of tiller number. PIN5b overexpression significantly decreases tiller number, whereas pin5b mutants show a greater tiller number (Lu et al. 2015). Notably, TaPIN5b was upregulated at both stages in ot1. Further research is required to determine whether this gene affects tiller growth in ot1.
Reduced tiller number in ot1 may not be controlled by currently known tillering genes
To identify genes controlling tiller development in ot1, we analyzed the expression patterns of known homeologs of tiller development genes in rice and wheat. HAP2E overexpression increases tiller number (Alam et al. 2015). PGL/CAO1 promotes axillary bud growth (Yang et al. 2016). MOC1 and FON1 promote tiller growth (Moon et al. 2006; Shao et al. 2019). These genes were significantly upregulated in ot1 and were more highly expressed at the jointing stage compared with the regreening stage. This indicated that the effects of these genes were gradually enhanced as tiller development progressed, but that they did not increase the tiller number.
OsbHLH025 and IPA1 overexpression decreases tiller number (Yamamura et al. 2015; Song et al. 2017), suggesting that repression of both genes would lead to a greater tiller number. We here found that both genes were significantly downregulated in the ot1 mutant at both stages. The tiller number, plant height, and spike length of ot1 plants were similar to those of tn1 mutants (Dong et al. 2023). However, TN1 expression was not altered in ot1 (Fig. 7c), meaning that the gene regulating oligo-tillering in ot1 differed from the one in tn1 and in other known tillering mutants.
Conclusions
We have found that tiller number was significantly decreased in ot1 mutants compared to the WT at the seedling stage, but that thousand-grain weight was significantly increased. ot1 had a distinct phenotype compared to other known wheat oligo-tillering mutants. RNA-seq of the tiller bud revealed upregulation of genes associated with SL and ABA biosynthesis and signaling. Tiller number inhibition in ot1 may therefore have been related to the activity of plant hormone pathways. Known tiller growth-promoting genes were significantly upregulated, whereas genes known to inhibit tiller development were downregulated in ot1. Thus, ot1 may reveal a novel locus for tiller number inhibition.
Data availability
All data generated or analyzed in this study are included in the manuscript and the supplementary information files. The RNA-seq data were deposited to the National Center for Biotechnology Information (NCBI) SRA repository under accession number PRJNA996360.
References
Abuauf H, Haider I, Jia K, Ablazov A, Mi J, Blilou I, Al-Babili S (2018) The Arabidopsis DWARF27 gene encodes an all-trans-/9-cis-β-carotene isomerase and is induced by auxin, abscisic acid and phosphate deficiency. Plant Sci 277:33–42. https://doi.org/10.1016/j.plantsci.2018.06.024
Ahmed HI, Heuberger M, Schoen A, Koo D, Quiroz-Chavez J, Adhikari L, Raupp J, Cauet S, Rodde N, Cravero C, Callot C, Lazo GR, Kathiresan N, Sharma PK, Moot I, Yadav IS, Singh L, Saripalli G, Rawat N, Datla R, Athiyannan N, Ramirez-Gonzalez RH, Uauy C, Wicker T, Tiwari VK, Abrouk M, Poland J, Krattinger SG (2023) Einkorn genomics sheds light on history of the oldest domesticated wheat. Nature 620(7975):830–838. https://doi.org/10.1038/s41586-023-06389-7
Alam MM, Tanaka T, Nakamura H, Ichikawa H, Kobayashi K, Yaeno T, Yamaoka N, Shimomoto K, Takayama K, Nishina H, Nishiguchi M (2015) Overexpression of a rice heme activator protein gene (OsHAP2E) confers resistance to pathogens, salinity and drought, and increases photosynthesis and tiller number. Plant Biotechnol J 13(1):85–96. https://doi.org/10.1111/pbi.12239
Al-Babili S, Bouwmeester HJ (2015) Strigolactones, a novel carotenoid-derived plant hormone. Annu Rev Plant Biol 66(1):161–186. https://doi.org/10.1146/annurev-arplant-043014-114759
Alder A, Jamil M, Marzorati M, Bruno M, Vermathen M, Bigler P, Ghisla S, Bouwmeester H, Beyer P, Al-Babili S (2012) The path from β-carotene to carlactone, a strigolactone-like plant hormone. Science 335(6074):1348–1351. https://doi.org/10.1126/science.1218094
An J, Niu H, Ni Y, Jiang Y, Zheng Y, He R, Li J, Jiao Z, Zhang J, Li H, Li Q, Niu J (2019) The miRNA–mRNA networks involving abnormal energy and hormone metabolisms restrict tillering in a wheat mutant dmc. Int J Mol Sci 20(18):4586. https://doi.org/10.3390/ijms20184586
Bang SW, Park S, Jeong JS, Kim YS, Jung H, Ha S, Kim J (2013) Characterization of the stress-inducible OsNCED3 promoter in different transgenic rice organs and over three homozygous generations. Planta 237(1):211–224. https://doi.org/10.1007/s00425-012-1764-1
Barrero JM, Piqueras P, González-Guzmán M, Serrano R, Rodríguez PL, Ponce MR, Micol JL (2005) A mutational analysis of the ABA1 gene of Arabidopsis thaliana highlights the involvement of ABA in vegetative development. J Exp Bot 56(418):2071–2083. https://doi.org/10.1093/jxb/eri206
Bhatnagar N, Min M, Choi E, Kim N, Moon S, Yoon I, Kwon T, Jung K, Kim B (2017) The protein phosphatase 2C clade A protein OsPP2C51 positively regulates seed germination by directly inactivating OsbZIP10. Plant Mol Biol 93(4):389–401. https://doi.org/10.1007/s11103-016-0568-2
Booker J, Sieberer T, Wright W, Williamson L, Willett B, Stirnberg P, Turnbull C, Srinivasan M, Goddard P, Leyser O (2005) MAX1 encodes a cytochrome P450 family member that acts downstream of MAX3/4 to produce a carotenoid-derived branch-inhibiting hormone. Dev Cell 8(3):443–449. https://doi.org/10.1016/j.devcel.2005.01.009
Bray NL, Pimentel H, Melsted P, Pachter L (2016) Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol 34(5):525–527. https://doi.org/10.1038/nbt.3519
Chen Y, Fan X, Song W, Zhang Y, Xu G (2012) Over-expression of OsPIN2 leads to increased tiller numbers, angle and shorter plant height through suppression of OsLAZY1. Plant Biotechnol J 10(2):139–149. https://doi.org/10.1111/j.1467-7652.2011.00637.x
Chen K, Li GJ, Bressan RA, Song CP, Zhu JK, Zhao Y (2020) Abscisic acid dynamics, signaling, and functions in plants. J Integr Plant Biol 62(1):25–54. https://doi.org/10.1111/jipb.12899
Dabbert T, Okagaki RJ, Cho S, Heinen S, Boddu J, Muehlbauer GJ (2010) The genetics of barley low-tillering mutants: low number of tillers-1 (lnt1). Theor Appl Genet 121(4):705–715. https://doi.org/10.1007/s00122-010-1342-5
Dall’Osto L, Cazzaniga S, North H, Marion-Poll A, Bassi R (2007) The Arabidopsis aba4-1 mutant reveals a specific function for neoxanthin in protection against photooxidative stress. Plant Cell 19(3):1048–1064. https://doi.org/10.1105/tpc.106.049114
Dong C, Zhang L, Zhang Q, Yang Y, Li D, Xie Z, Cui G, Chen Y, Wu L, Li Z, Liu G, Zhang X, Liu C, Chu J, Zhao G, Xia C, Jia J, Sun J, Kong X, Liu X (2023) Tiller Number1 encodes an ankyrin repeat protein that controls tillering in bread wheat. Nat Commun 14(1):836–849. https://doi.org/10.1038/s41467-023-36271-z
Duan J, Yu H, Yuan K, Liao Z, Meng X, Jing Y, Liu G, Chu J, Li J (2019) Strigolactone promotes cytokinin degradation through transcriptional activation of Cytokinin Oxidase/Dehydrogenase 9 in rice. Proc Natl Acad Sci 116(28):14319–14324. https://doi.org/10.1073/pnas.1810980116
Fiorilli V, Wang JY, Bonfante P, Lanfranco L, Al-Babili S (2019) Apocarotenoids: old and new mediators of the arbuscular mycorrhizal symbiosis. Front Plant Sci 10:1186. https://doi.org/10.3389/fpls.2019.01186
Gomez-Roldan V, Fermas S, Brewer PB, Puech-Pagès V, Dun EA, Pillot J, Letisse F, Matusova R, Danoun S, Portais J, Bouwmeester H, Bécard G, Beveridge CA, Rameau C, Rochange SF (2008) Strigolactone inhibition of shoot branching. Nature 455(7210):189–194. https://doi.org/10.1038/nature07271
González-Grandío E, Pajoro A, Franco-Zorrilla JM, Tarancón C, Immink RGH, Cubas P (2017) Abscisic acid signaling is controlled by a BRANCHED1/HD-ZIP I cascade in Arabidopsis axillary buds. Proc Natl Acad Sci 114(2):E245–E254. https://doi.org/10.1073/pnas.1613199114
Guo S, Xu Y, Liu H, Mao Z, Zhang C, Ma Y, Zhang Q, Meng Z, Chong K (2013) The interaction between OsMADS57 and OsTB1 modulates rice tillering via DWARF14. Nat Commun 4(1):1566–1578. https://doi.org/10.1038/ncomms2542
Guo H, Yan Z, Li X, Xie Y, Xiong H, Liu Y, Zhao L, Gu J, Zhao S, Liu L (2017) Development of a high-efficient mutation resource with phenotypic variation in hexaploid winter wheat and identification of novel alleles in the TaAGP.L-B1 gene. Front Plant Sci 8:1404. https://doi.org/10.3389/fpls.2017.01404
Hou M, Luo F, Wu D, Zhang X, Lou M, Shen D, Yan M, Mao C, Fan X, Xu G, Zhang Y (2021) OsPIN9, an auxin efflux carrier, is required for the regulation of rice tiller bud outgrowth by ammonium. New Phytol 229(2):935–949. https://doi.org/10.1111/nph.16901
Ishikawa S, Maekawa M, Arite T, Onishi K, Takamure I, Kyozuka J (2005) Suppression of tiller bud activity in tillering dwarf mutants of rice. Plant Cell Physiol 46(1):79–86. https://doi.org/10.1093/pcp/pci022
Ito S, Braguy J, Wang JY, Yoda A, Fiorilli V, Takahashi I, Jamil M, Felemban A, Miyazaki S, Mazzarella T, Chen GE, Shinozawa A, Balakrishna A, Berqdar L, Rajan C, Ali S, Haider I, Sasaki Y, Yajima S, Akiyama K, Lanfranco L, Zurbriggen MD, Nomura T, Asami T, Al-Babili S (2022) Canonical strigolactones are not the major determinant of tillering but important rhizospheric signals in rice. Sci Adv 8(44):d1278. https://doi.org/10.1126/sciadv.add1278
Jiang L, Liu X, Xiong G, Liu H, Chen F, Wang L, Meng X, Liu G, Yu H, Yuan Y, Yi W, Zhao L, Ma H, He Y, Wu Z, Melcher K, Qian Q, Xu HE, Wang Y, Li J (2013) DWARF 53 acts as a repressor of strigolactone signalling in rice. Nature 504(7480):401–405. https://doi.org/10.1038/nature12870
Kapulnik Y, Delaux P, Resnick N, Mayzlish-Gati E, Wininger S, Bhattacharya C, Séjalon-Delmas N, Combier J, Bécard G, Belausov E, Beeckman T, Dor E, Hershenhorn J, Koltai H (2011) Strigolactones affect lateral root formation and root-hair elongation in Arabidopsis. Planta 233(1):209–216. https://doi.org/10.1007/s00425-010-1310-y
Kebrom TH, Chandler PM, Swain SM, King RW, Richards RA, Spielmeyer W (2012) Inhibition of tiller bud outgrowth in the tin mutant of wheat is associated with precocious internode development. Plant Physiol 160(1):308–318. https://doi.org/10.1104/pp.112.197954
Kim H, Hwang H, Hong J, Lee Y, Ahn IP, Yoon IS, Yoo S, Lee S, Lee SC, Kim B (2012) A rice orthologue of the ABA receptor, OsPYL/RCAR5, is a positive regulator of the ABA signal transduction pathway in seed germination and early seedling growth. J Exp Bot 63(2):1013–1024. https://doi.org/10.1093/jxb/err338
Kuraparthy V, Sood S, Dhaliwal HS, Chhuneja P, Gill BS (2006) Identification and mapping of a tiller inhibition gene (tin3) in wheat. Theor Appl Genet 114(2):285–294. https://doi.org/10.1007/s00122-006-0431-y
Li X, Qian Q, Fu Z, Wang Y, Xiong G, Zeng D, Wang X, Liu X, Teng S, Hiroshi F, Yuan M, Luo D, Han B, Li J (2003) Control of tillering in rice. Nature 422(6932):618–621. https://doi.org/10.1038/nature01518
Liu X, Hu Q, Yan J, Sun K, Liang Y, Jia M, Meng X, Fang S, Wang Y, Jing Y, Liu G, Wu D, Chu C, Smith SM, Chu J, Wang Y, Li J, Wang B (2020) ζ-Carotene isomerase suppresses tillering in rice through the coordinated biosynthesis of strigolactone and abscisic acid. Mol Plant 13(12):1784–1801. https://doi.org/10.1016/j.molp.2020.10.001
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 25(4):402–408. https://doi.org/10.1006/meth.2001.1262
Lu G, Coneva V, Casaretto JA, Ying S, Mahmood K, Liu F, Nambara E, Bi YM, Rothstein SJ (2015) OsPIN5b modulates rice (Oryza sativa) plant architecture and yield by changing auxin homeostasis, transport and distribution. Plant J 83(5):913–925. https://doi.org/10.1111/tpj.12939
Luo Z, Janssen BJ, Snowden KC (2021) The molecular and genetic regulation of shoot branching. Plant Physiol 187(3):1033–1044. https://doi.org/10.1093/plphys/kiab071
Ma B, Yin C, He S, Lu X, Zhang W, Lu T, Chen S, Zhang J (2014) Ethylene-induced inhibition of root growth requires abscisic acid function in rice (Oryza sativa L.) seedlings. Plos Genet 10(10):e1004701
Mitchell JH, Rebetzke GJ, Chapman SC, Fukai S (2013) Evaluation of reduced-tillering (tin) wheat lines in managed, terminal water deficit environments. J Exp Bot 64(11):3439–3451. https://doi.org/10.1093/jxb/ert181
Moon S, Jung KH, Lee DE, Lee DY, Lee J, An K, Kang HG, An G (2006) The rice FON1 gene controls vegetative and reproductive development by regulating shoot apical meristem size. Mol Cells 21(1):147–152
Nambara E, Kawaide H, Kamiya Y, Naito S (1998) Characterization of an Arabidopsis thaliana mutant that has a defect in ABA accumulation: ABA-dependent and ABA-independent accumulation of free amino acids during dehydration. Plant Cell Physiol 39(8):853–858. https://doi.org/10.1093/oxfordjournals.pcp.a029444
Oikawa T, Kyozuka J (2009) Two-Step regulation of LAX PANICLE1 protein accumulation in axillary meristem formation in rice. Plant Cell 21(4):1095–1108. https://doi.org/10.1105/tpc.108.065425
Peng Z, Yen C, Yang JL (1998) Genetic control of oligo-culms in common wheat. Wheat Inf Serv 26:19–24
Richards RA (1988) A tiller inhibitor gene in wheat and its effect on plant growth. Aust J AGR RES 39(5):749–757. https://doi.org/10.1071/AR9880749
Robinson MD, McCarthy DJ, Smyth GK (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26(1):139–140. https://doi.org/10.1093/bioinformatics/btp616
Schoen A, Yadav I, Wu S, Poland J, Rawat N, Tiwari V (2023) Identification and high-resolution mapping of a novel tiller number gene (tin6) by combining forward genetics screen and MutMap approach in bread wheat. Funct Integr Genomic 23(2):157. https://doi.org/10.1007/s10142-023-01084-2
Shabek N, Ticchiarelli F, Mao H, Hinds TR, Leyser O, Zheng N (2018) Structural plasticity of D3–D14 ubiquitin ligase in strigolactone signalling. Nature 563(7733):652–656. https://doi.org/10.1038/s41586-018-0743-5
Shang Q, Wang Y, Tang H, Sui N, Zhang X, Wang F (2021) Genetic, hormonal, and environmental control of tillering in wheat. Crop J 9(5):986–991. https://doi.org/10.1016/j.cj.2021.03.002
Shao G, Lu Z, Xiong J, Wang B, Jing Y, Meng X, Liu G, Ma H, Liang Y, Chen F, Wang Y, Li J, Yu H (2019) Tiller bud formation regulators MOC1 and MOC3 cooperatively promote tiller bud outgrowth by activating FON1 expression in rice. Mol Plant 12(8):1090–1102. https://doi.org/10.1016/j.molp.2019.04.008
Shiferaw B, Smale M, Braun H, Duveiller E, Reynolds M, Muricho G (2013) Crops that feed the world 10. Past successes and future challenges to the role played by wheat in global food security. Food Secur 5(3):291–317. https://doi.org/10.1007/s12571-013-0263-y
Si Y, Lu Q, Tian S, Niu J, Cui M, Liu X, Gao Q, Shi X, Ling H, Zheng S (2022) Fine mapping of the tiller inhibition gene TIN5 in Triticum urartu. Theor Appl Genet 135(8):2665–2673. https://doi.org/10.1007/s00122-022-04140-w
Song X, Lu Z, Yu H, Shao G, Xiong J, Meng X, Jing Y, Liu G, Xiong G, Duan J, Yao X, Liu C, Li H, Wang Y, Li J (2017) IPA1 functions as a downstream transcription factor repressed by D53 in strigolactone signaling in rice. Cell Res 27(9):1128–1141. https://doi.org/10.1038/cr.2017.102
Sun H, Tao J, Gu P, Xu G, Zhang Y (2016) The role of strigolactones in root development. Plant Signal Behav 11(1):e1110662. https://doi.org/10.1080/15592324.2015.1110662
Tabuchi H, Zhang Y, Hattori S, Omae M, Shimizu-Sato S, Oikawa T, Qian Q, Nishimura M, Kitano H, Xie H, Fang X, Yoshida H, Kyozuka J, Chen F, Sato Y (2011) LAX PANICLE2 of rice encodes a novel nuclear protein and regulates the formation of axillary meristems. Plant Cell 23(9):3276–3287. https://doi.org/10.1105/tpc.111.088765
Takeda T, Suwa Y, Suzuki M, Kitano H, Ueguchi-Tanaka M, Ashikari M, Matsuoka M, Ueguchi C (2003) The OsTB1 gene negatively regulates lateral branching in rice. Plant J 33(3):513–520. https://doi.org/10.1046/j.1365-313x.2003.01648.x
Tal L, Palayam M, Ron M, Young A, Britt A, Shabek N (2022) A conformational switch in the SCF-D3/MAX2 ubiquitin ligase facilitates strigolactone signalling. Nat Plants. https://doi.org/10.1038/s41477-022-01145-7
Wang Z, Wu F, Chen X, Zhou W, Shi H, Lin Y, Hou S, Yu S, Zhou H, Li C, Liu Y (2022) Fine mapping of the tiller inhibition gene TIN4 contributing to ideal plant architecture in common wheat. Theor Appl Genet 135(2):527–535. https://doi.org/10.1007/s00122-021-03981-1
Watanabe S, Sato M, Sawada Y, Tanaka M, Matsui A, Kanno Y, Hirai MY, Seki M, Sakamoto A, Seo M (2018) Arabidopsis molybdenum cofactor sulfurase ABA3 contributes to anthocyanin accumulation and oxidative stress tolerance in ABA-dependent and independent ways. Sci Rep 8(1):16592. https://doi.org/10.1038/s41598-018-34862-1
Xie C, Mao X, Huang J, Ding Y, Wu J, Dong S, Kong L, Gao G, Li CY, Wei L (2011) KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res 39(Web Server issue):W316–W322. https://doi.org/10.1093/nar/gkr483
Yamamura C, Mizutani E, Okada K, Nakagawa H, Fukushima S, Tanaka A, Maeda S, Kamakura T, Yamane H, Takatsuji H, Mori M (2015) Diterpenoid phytoalexin factor, a bHLH transcription factor, plays a central role in the biosynthesis of diterpenoid phytoalexins in rice. Plant J 84(6):1100–1113. https://doi.org/10.1111/tpj.13065
Yang Y, Xu J, Huang L, Leng Y, Dai L, Rao Y, Chen L, Wang Y, Tu Z, Hu J, Ren D, Zhang G, Zhu L, Guo L, Qian Q, Zeng D (2016) PGL, encoding chlorophyllide a oxygenase 1, impacts leaf senescence and indirectly affects grain yield and quality in rice. J Exp Bot 67(5):1297–1310. https://doi.org/10.1093/jxb/erv529
Yao C, Finlayson SA (2015) Abscisic acid is a general negative regulator of Arabidopsis axillary bud growth. Plant Physiol 169(1):611–626. https://doi.org/10.1104/pp.15.00682
Yoneyama K, Xie X, Yoneyama K, Kisugi T, Nomura T, Nakatani Y, Akiyama K, McErlean CSP (2018) Which are the major players, canonical or non-canonical strigolactones? J Exp Bot 69(9):2231–2239. https://doi.org/10.1093/jxb/ery090
Young MD, Wakefield MJ, Smyth GK, Oshlack A (2010) Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol 11(2):R14. https://doi.org/10.1186/gb-2010-11-2-r14
Zhang J, Wu J, Liu W, Lu X, Yang X, Gao A, Li X, Lu Y, Li L (2013) Genetic mapping of a fertile tiller inhibition gene, ftin, in wheat. Mol Breeding 31(2):441–449. https://doi.org/10.1007/s11032-012-9801-0
Zhang Y, van Dijk ADJ, Scaffidi A, Flematti GR, Hofmann M, Charnikhova T, Verstappen F, Hepworth J, van der Krol S, Leyser O, Smith SM, Zwanenburg B, Al-Babili S, Ruyter-Spira C, Bouwmeester HJ (2014) Rice cytochrome P450 MAX1 homologs catalyze distinct steps in strigolactone biosynthesis. Nat Chem Biol 10(12):1028–1033. https://doi.org/10.1038/nchembio.1660
Zhao L, Zhou XE, Wu Z, Yi W, Xu Y, Li S, Xu T, Liu Y, Chen R, Kovach A, Kang Y, Hou L, He Y, Xie C, Song W, Zhong D, Xu Y, Wang Y, Li J, Zhang C, Melcher K, Xu HE (2013) Crystal structures of two phytohormone signal-transducing α/β hydrolases: karrikin-signaling KAI2 and strigolactone-signaling DWARF14. Cell Res 23(3):436–439. https://doi.org/10.1038/cr.2013.19
Zhao B, Wu TT, Ma SS, Jiang DJ, Bie XM, Sui N, Zhang XS, Wang F (2019) TaD27-B gene controls the tiller number in hexaploid wheat. Plant Biotechnol J 18(2):513–525. https://doi.org/10.1111/pbi.13220
Zhou F, Lin Q, Zhu L, Ren Y, Zhou K, Shabek N, Wu F, Mao H, Dong W, Gan L, Ma W, Gao H, Chen J, Yang C, Wang D, Tan J, Zhang X, Guo X, Wang J, Jiang L, Liu X, Chen W, Chu J, Yan C, Ueno K, Ito S, Asami T, Cheng Z, Wang J, Lei C, Zhai H, Wu C, Wang H, Zheng N, Wan J (2013) D14–SCFD3-dependent degradation of D53 regulates strigolactone signalling. Nature 504(7480):406–410. https://doi.org/10.1038/nature12878
Zhu G, Ye N, Zhang J (2009) Glucose-induced delay of seed germination in rice is mediated by the suppression of ABA catabolism rather than an enhancement of ABA biosynthesis. Plant Cell Physiol 50(3):644–651. https://doi.org/10.1093/pcp/pcp022
Funding
This work was supported by the National Key Research and Development Project of China (2022YFD1200700) and the China Agriculture Research System CARS-03.
Author information
Authors and Affiliations
Contributions
JB and HG wrote the manuscript. JB, HX, YX, JG, LZ, SZ, and YD performed the experiments and analyzed the data. LL and HG designed the experiment. All authors have read and approved the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bai, J., Guo, H., Xiong, H. et al. Strigolactone and abscisic acid synthesis and signaling pathways are enhanced in the wheat oligo-tillering mutant ot1. Mol Breeding 44, 12 (2024). https://doi.org/10.1007/s11032-024-01450-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11032-024-01450-3