
Abstract
Cardiovascular diseases (CVD) are closely linked to protein homeostasis (proteostasis) and its failure. Beside genetic mutations that impair cardiac protein quality control, obesity is a strong risk factor for heart disease. In obesity, adipose tissue becomes dysfunctional and impacts heart function and CVD progression by releasing cytokines that contribute to systemic insulin resistance and cardiovascular dysfunction. In addition, chronic inflammation and lipotoxicity compromise endoplasmic reticulum (ER) function, eliciting stress responses that overwhelm protein quality control beyond its capacity. Impairment of proteostasis—including dysfunction of the ubiquitin–proteasome system (UPS), autophagy, and the depletion of chaperones—is intricately linked to cardiomyocyte dysfunction. Interventions targeting UPS and autophagy pathways are new potential strategies for re-establishing protein homeostasis and improving heart function. Additionally, lifestyle modifications such as dietary interventions and exercise have been shown to promote cardiac proteostasis and overall metabolic health. The pursuit of future research dedicated to proteostasis and protein quality control represents a pioneering approach for enhancing cardiac health and addressing the complexities of obesity-related cardiac dysfunction.
Zusammenfassung
Kardiovaskuläre Erkrankungen stehen in enger Verbindung mit der Proteinhomöostase (Proteostase) und deren Kollaps. Neben genetischen Mutationen, welche die Qualitätssicherung von kardialen Proteinen beeinträchtigen, stellt Adipositas einen starken Risikofaktor für Herzerkrankungen dar. Bei Adipositas liegt eine Fehlfunktion des Fettgewebes vor, welche die Herzfunktion sowie den Verlauf von kardiovaskulären Erkrankungen beeinflusst, indem das Fettgewebe Zytokine freisetzt, die zur systemischen Insulinresistenz und zu kardiovaskulären Dysfunktionen beitragen. Darüber hinaus beeinträchtigen chronische Entzündung und Lipotoxizität die Funktion des endoplasmatischen Retikulums (ER) und lösen Stressreaktionen aus, welche die Qualitätskontrolle von Proteinen beeinträchtigen. Eine gestörte Proteostase, einschließlich der Dysfunktion des Ubiquitin-Proteasom-Systems (UPS), der Autophagie und des Verlusts von Chaperonen, ist eng mit der Dysfunktion von Kardiomyozyten verbunden. Interventionen, die mittels UPS- und Autophagie darauf abzielen, die Proteinhomöostase wiederherzustellen, sind potenziell neue Strategien, um die Herzfunktion zu verbessern. Anpassungen des Lebensstils, eine Ernährungsumstellung und verstärkte Bewegung haben sich als unterstützend für die kardiale Proteostase und die allgemeine Stoffwechselgesundheit erwiesen. Das Betreiben künftiger Forschung auf dem Gebiet der Proteostase und der Qualitätskontrolle von Proteinen stellt potenziell einen bahnbrechenden Ansatz zur Verbesserung der kardialen Gesundheit und zur Erfassung der Komplexität von durch Adipositas bedingten Herzkrankheiten dar.
Similar content being viewed by others


Obesity is a disease of excess dysfunctional adipose tissue defined by a body mass index (BMI) of 30 kg/m2 or higher and is closely linked to a plethora of comorbidities including type 2 diabetes and cardiovascular disease (CVD). The impact of obesity on the cardiovascular system remains particularly concerning, as obesity induces metabolic and inflammatory changes, increasing the risk for life-threatening arrhythmias, atherosclerosis, and thrombosis [1].
At the molecular level, the concept of proteostasis—regulated homeostasis of protein metabolism—is emerging as a critical aspect in the context of cardiometabolic diseases. Cellular proteostasis is maintained by a tightly regulated network of molecular chaperones, proteases, and various quality control systems that collectively oversee protein translation, folding, and degradation [2]. Proteostasis is highly adaptive and indispensable for cellular function. However, in obesity, factors such as lipotoxicity, oxidative stress, and chronic inflammation impair this delicate balance, leading to the accumulation of misfolded and damaged proteins, particularly detrimental to cardiac structures and function [3]. New therapeutic approaches aiming at restoring proteostasis, including heat shock proteins and proteasome inhibitors, as well as non-pharmacological strategies such as dietary restriction and exercise, may have the potential to improve obesity-associated comorbidities. This review discusses the complex relationship between obesity, CVD, and proteostasis, highlighting the underlying basic biology, pathophysiological mechanisms, and therapeutic strategies.
Obesity-induced dysfunction of the heart
Obesity, characterized by dysfunctional adipose tissue, is an endocrine disorder associated with a systemic inflammatory state and cardiovascular consequences. During excessive weight gain, adipose tissue undergoes a transformation from being a dedicated and safe lipid storage organ with a favorable endocrine profile to a major site of tissue inflammation and detrimental cytokine secretion. Adipocytes secrete various signaling molecules and cytokines such as leptin, adiponectin, and resistin, which normally maintain metabolic balance. However, in obesity, adipocytes become hypertrophic and the secretome changes, directly contributing to weight gain, metabolic stress, and systemic inflammation [4, 5]. The escalation of non-esterified fatty acid release from hypertrophic adipocytes further exacerbates metabolic stress by promoting systemic insulin resistance and ectopic lipid deposition in non-adipose tissues such as the liver, muscle, and particularly the heart, setting the stage for cardiovascular dysfunction [6]. In the cardiovascular system, the sequelae of obesity also manifest as altered hemodynamics, such as increased cardiac output, heightened systemic vascular resistance, and obesity-induced hypertension. This link between metabolic disruption and cardiovascular health is causal, as the direct impact of obesity on cardiac structure and function is evident, alongside the indirect effects mediated by obesity-associated comorbidities [1].
Lipotoxicity, a consequence of obesity, is characterized by the harmful accumulation of lipids within non-adipose tissues, including the heart, where they harm myocardial function. This lipotoxic environment prompts myocardial injury and triggers maladaptive cardiac remodeling, manifesting as fibrosis and cardiomyopathy, and compromising both diastolic and systolic heart functions [7]. Coupled with abnormal lipid profiles, obesity-related stress on cardiomyocytes, attributed in part to disturbed proteostasis, contributes to a deleterious cycle. Disrupted endoplasmic reticulum (ER) function induces a stress response that, when overwhelmed, leads to proteostasis imbalance, which is integral to maintaining cell viability and function. Given the robust epidemiological evidence linking obesity and CVD, a deeper understanding of the roles of ER stress and proteostasis in this interplay is warranted.
Activation of the integrated stress response
Chronic metabolic overload and inflammatory stress disrupt ER function, an organelle pivotal in coordinating cellular metabolism and stress responses [8, 9]. The role of the ER in lipid and protein homeostasis is especially critical in cardiomyocytes, which demand precise coordination of protein synthesis and folding to sustain continuous cardiac function. Endoplasmic reticulum stress triggers the integrated stress response (ISR), including the unfolded protein response (UPR), ER-associated degradation (ERAD), and the ubiquitin-proteasome system (UPS). This preserves proteostasis, enhancing protein folding capabilities, regulating translation, and facilitating the targeted degradation of damaged proteins [10]. The role of the ER extends to regulate lipid synthesis and droplet formation, crucial for energy storage and utilization [11]. Furthermore, ER stress-induced lipotoxicity exacerbates systemic metabolic dysfunction, culminating in whole-body insulin resistance and, consequently, increased risk of developing CVD [7]. It is also noteworthy to mention that ER stress influences mitochondrial function through direct and indirect mechanisms. The ER and mitochondria are physically and functionally connected through structures known as mitochondria-associated ER membranes (MAMs). Disruptions in the integrity of MAMs lead to mitochondrial dysfunction, further worsening metabolic and cardiovascular outcomes [12].
Autophagy is another line of defense against ER stress in cardiomyocytes, orchestrating the degradation of protein aggregates to preserve cellular health. Autophagy is a multi-step cellular recycling process, initiated through the formation of a phagophore, a membrane that sequesters damaged proteins and organelles. These autophagosomes migrate and fuse with lysosomes, breaking down complex proteins into amino acids and other basic components [13]. The resulting macromolecular breakdown products are then recycled, ready to be repurposed for the synthesis of new proteins or to be utilized for energy production. The integrity of this system is especially vital in the context of cardiac physiology. During states of metabolic stress, such as those induced by obesity, the autophagic-lysosomal pathway is upregulated as a compensatory mechanism. It helps mitigate the accumulation of misfolded proteins that disrupt cardiac function, potentially leading to conditions such as cardiomyopathy and heart failure [14].
Maladaptation of cardiac proteostasis
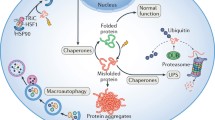
Cardiomyocytes, with their specialized roles in electrical conduction and contraction, rely on the integrity of the proteome for optimal heart function. Due to the high metabolic demand of these cells, maintaining proteostasis presents unique challenges not commonly encountered by other cell types [15, 16]. Functional proteostasis is essential for a healthy heart, whereas proteome imbalances, such as misfolded proteins and aggregates, are hallmarks of cardiac disease (Fig. 1). The protein quality control systems, including the UPS and autophagy pathways, are critical in eliminating deleterious proteins and maintaining cellular integrity [17]. These systems are orchestrated by the UPR and ERAD. The UPR sensors, such as IRE‑1, PERK, and ATF6, respond to ER stress by activating adaptive pathways to restore proteostasis [18]. They also serve to rewire metabolism, as recently shown in a mouse model for Barth syndrome, in which activation of ATF4 compensated for defects in mitochondrial uptake of fatty acids to sustain energy production and antioxidation [19]. However, when overwhelmed, these sensors may trigger maladaptive responses, potentially leading to cardiac pathology as outlined here (Fig. 1).

Cardiac proteostasis and its adaptation and dysfunction. Proteostasis describes a state of homeostatic protein production and degradation, usually found in healthy individuals, stimulated by healthy diets and physical activity. The occurrence of proteotoxic stress, e.g., presence of misfolded proteins and damaged proteins or simply caused by changes in proteome turnover are mitigated by the adaptive activation of the integrated stress response to restore homeostasis. If the proteotoxic stress persists, the cell enters a maladaptive phase characterized by organelle stress, inflammation, and fibrosis, which ultimately lead to cardiac dysfunction. Risk factors include aging, metabolic syndrome, and genetic predispositions. UPR unfolded protein response, UPS ubiquitin-proteasome system
The UPS is particularly sensitive to metabolic conditions such as obesity. Disruptions in UPS, exemplified by studies such as those involving PA28alpha (a proteasome activator) knockout mice, lead to the accumulation of polyubiquitinated proteins and desmin-related cardiomyopathy [20]. Unresolved chronic ER stress contributes to this by accumulating partially unfolded or misfolded proteins. Despite UPS activation in response to obesity and heart dysfunction, its capacity to mitigate proteotoxic stress may be inadequate or even maladaptive. Some of the detrimental outcomes of proteasome dysfunction seem to be linked to activation of ATF3 [21]. A key regulator of fine-tuning proteasome function and UPS is the NFE2-like bZIP transcription factor 1 (NFE2L1), a transcription factor that regulates the expression of genes coding of proteasome subunits and components [22, 23]. In the absence of stress, NFE2L1 is continuously degraded by a cascade of events involving deglycosylation by NGLY1 and proteolytic cleavage by DDI2 [24, 25]. However, during proteotoxic stress, for example, in the presence of chemical proteasome inhibitors or high levels of ubiquitylated proteins, NFE2L1 evades degradation and upregulates proteasome subunit production to counteract the stress. Deficiency of NFE2L1 has been linked to several physiologic and pathologic conditions, including compromised tissue regeneration and response to injury [26,27,28]. We and others have shown that loss of NFE2L1 is linked to ferroptosis, a cell death modality that involves lipid reactive oxygen species (ROS; [29]). Conversely, NFE2L1 overexpression enhances cardiac repair and protects against ischemia/reperfusion injury [28].
In humans, Predmore et al. reported reduced UPS activity in cardiac tissue samples obtained from patients with heart failure or hypertrophic cardiomyopathy, providing clinical evidence of proteostasis dysfunction in these heart diseases [30]. Furthermore, in a prospective study conducted by Makris et al., treatment with clinical proteasome inhibitors was associated with deteriorated cardiac function, reinforcing the significance of UPS in maintaining cardiac health. The study revealed left ventricular dysfunction and a deterioration of left atrial remodeling in these patients [31]. Heart diseases such as atrial fibrillation show impaired proteasome function, where sustained cardiomyocyte stress leads to dysfunctional autophagy and protease activity, resulting in contractile and electrophysiological deficits. This impaired proteostasis, observed in conditions of cardiac hypertrophy due to pressure overload, manifests as autophagic degradation and protein aggregation when these degradation systems are overwhelmed [32]. Moreover, a study of a population in southern Taiwan illustrated a notable decline in plasma ubiquitin and 20S proteasome levels in obese individuals compared to normal controls [33]. This intersection of UPR, ERAD dysfunction, and the resulting disruption of proteostasis are evident in progressive CVD and offer potential biomarkers for assessing obesity-related cardiovascular risk.
Genetic impact on cardiac proteostasis
Cardiomyopathies are not only shaped by lifestyle factors but also by a spectrum of genetic mutations that directly influence cardiomyocytes proteostasis. Mutations in genes coding for valosin-containing protein (VCP) and BCL2-associated athanogene 3 (BAG3) primarily impact the proteasome. These VCP mutations lead to aberrant protein and RNA quality control, contributing to multisystem proteopathy with cardiac involvement [34]. The BAG3 mutations are implicated in dilated cardiomyopathy, affecting the chaperone-assisted selective autophagy (CASA) complex, crucial for proteasomal degradation of mechanically strained proteins in cardiomyocytes [35].
Lysosomal-associated membrane protein‑2 (LAMP2) mutations result in Danon disease, characterized by lysosomal dysfunction and impaired autophagy, often presenting with hypertrophic cardiomyopathy [36]. The disruption of autophagic processes is a common theme in cardiomyopathies, highlighting the critical role of lysosomal degradation pathways in cardiac proteostasis. Dystrophin (DMD) gene mutations, causing Duchenne muscular dystrophy, lead to compromised sarcolemma integrity and secondary proteostasis imbalance, culminating in dilated cardiomyopathy [37]. Similarly, titin (TTN) truncating mutations disrupt sarcomere function due to the role of titin as a molecular spring, essential for the mechanical stability and proteostasis of cardiomyocytes [38]. Genetic variants in alpha-actinin‑2 (ACTN2) are associated with several forms of cardiomyopathy, featuring protein aggregation, hypertrophy, myofibrillar disarray, and activation of both the ubiquitin-proteasome system and the autophagy-lysosomal pathway [39]. Filamin C (FLNC) mutations lead to diverse cardiomyopathic outcomes. Aggregation-prone mutations in FLNC result in hypertrophic cardiomyopathy or myofibrillar myopathies, whereas mutations leading to haploinsufficiency are mainly associated with dilated cardiomyopathy and cardiac arrhythmias [40]. Also, these genetic factors delineate a complex landscape where the precise regulation of protein turnover is fundamental for cardiac function. Collectively, these clinical observations and experimental data converge to highlight the critical association between UPS and cardiomyopathies.
Future directions in cardiovascular proteostasis management
Several therapeutic agents are in development that target protein quality control pathways, presenting new avenues for treating cardiomyopathies, including UPS and the autophagy pathway. While proteasome inhibition in cancer therapy is associated with cardiovascular side effects, there is potential for therapeutic applications in certain cardiac conditions, as restoring dystrophin complexes offers a new therapeutic strategy for muscular dystrophies such as Duchenne muscular dystrophy. The ability of the proteasome inhibitor bortezomib to mitigate progressive muscle degeneration could provide a significant benefit in preventing development of dilated cardiomyopathy and heart failure [41].
Traditional antimalarials, chloroquine and hydroxychloroquine, known for their anti-inflammatory effects, are being repurposed to inhibit autophagy, potentially mitigating autophagic vacuole accumulation in lysosomal disorders like Danon disease, where dysfunctional autophagy leads to pathological cardiac hypertrophy [42]. Conversely, spermidine has captured attention as an autophagy activator, with its selective induction of autophagy considered crucial for the clearance of aggregated proteins in cardiomyocytes, potentially improving cardiac outcomes in cardiomyopathies [43]. Additionally, molecules designed to enhance UPS function, such as USP14 inhibitors, are being developed to expedite the degradation of misfolded proteins, aiming to restore proteostasis [44].
Transthyretin stabilizers such as tafamidis and diflunisal present a novel approach to prevent the misfolding and aggregation of proteins implicated in amyloid cardiomyopathy [45]. Complementary to this, the chemical chaperone 4‑phenylbutyrate (4-PBA) is explored for its ability to enhance protein folding, potentially reducing the cardiac burden of proteotoxic stress [46]. Additionally, geranylgeranylacetone (GGA) is being studied for its capacity to induce heat shock proteins, a natural defense against misfolded proteins, offering protection in the context of cardiac proteostasis [47]. Tauroursodeoxycholic acid (TUDCA), with its chemical chaperone activity, is of interest for its potential to alleviate ER stress and modulate apoptotic pathways, which are critical in cardiomyocyte viability [48].
Complementing these pharmacological interventions, lifestyle modifications such as dietary restriction and regular exercise have been substantiated to beneficially modulate proteostasis. Dietary restriction, characterized by a controlled reduction in caloric intake that avoids malnutrition, orchestrates a cellular stress response pivotal for bolstering proteostasis. This adaptive response involves the upregulation of molecular chaperones that facilitate accurate protein folding, alongside the activation of proteolytic pathways crucial for the clearance of misfolded and potentially toxic proteins, thereby preventing protein aggregation and maintaining cellular homeostasis [49]. Complementarily, physical exercise exerts a cardioprotective effect by fostering adaptations of the cardiovascular system. One of the key mechanisms underlying this benefit is the activation of autophagy. Exercise-induced autophagy has been linked to enhanced cardiac function, attenuation of oxidative stress, and a deceleration of the age-associated decline in proteostasis, highlighting its role in the preservation of cardiac integrity [50]. Together, these strategies illustrate a concerted, pathway-oriented effort to combat cardiac diseases, spotlighting the integral role of protein quality control in the pathogenesis and treatment of cardiomyopathies.
Conclusion
Obesity-induced heart disease represents a spectrum of immunometabolic cellular changes linked to increased risk for cardiovascular disease (CVD). This underscores the need for a deeper understanding of the nuanced mechanisms contributing to obesity-related cardiovascular complications, especially as the prevalence of obesity rises globally among aging populations. At the heart of these mechanisms lies proteostasis, the delicate equilibrium of protein synthesis, folding, and degradation, which is essential for cardiac function. Perturbations in this system are increasingly recognized as precipitating factors in the development and progression of cardiomyopathies, not only tied to obesity but as a generalizable key element of heart biology. In light of these findings, it will be relevant to intensify research into novel interventions that bolster proteostasis. As we advance our understanding of proteostasis in the context of cardiac health, the potential to devise more effective treatments for obesity-related CVD becomes increasingly tangible.
References
Ortega FB, Lavie CJ, Blair SN (2016) Obesity and cardiovascular disease. Circ Res 118:1752–1770. https://doi.org/10.1161/CIRCRESAHA.115.306883
Jayaraj GG, Hipp MS, Hartl FU (2020) Functional modules of the proteostasis network. Cold Spring Harb Perspect Biol. https://doi.org/10.1101/cshperspect.a033951
Bartelt A, Widenmaier SB (2020) Proteostasis in thermogenesis and obesity. Biol Chem 401:1019–1030. https://doi.org/10.1515/hsz-2019-0427
Giroud M, Jodeleit H, Prentice KJ, Bartelt A (2022) Adipocyte function and the development of cardiometabolic disease. J Physiol 600:1189–1208. https://doi.org/10.1113/JP281979
Reilly SM, Saltiel AR (2017) Adapting to obesity with adipose tissue inflammation. Nat Rev Endocrinol 13:633–643. https://doi.org/10.1038/nrendo.2017.90
Koenen M, Hill MA, Cohen P, Sowers JR (2021) Obesity, adipose tissue and vascular dysfunction. Circ Res 128:951–968. https://doi.org/10.1161/CIRCRESAHA.121.318093
Shreya S, Alam MJ, Anupriya A et al (2023) Lipotoxicity, ER stress, and cardiovascular disease: current understanding and future directions. Cardiovasc Hematol Agents Med Chem. https://doi.org/10.2174/0118715257262366230928051902
Arruda AP, Hotamisligil GS (2015) Calcium homeostasis and organelle function in the pathogenesis of obesity and diabetes. Cell Metab 22:381–397. https://doi.org/10.1016/j.cmet.2015.06.010
Lemmer IL, Willemsen N, Hilal N, Bartelt A (2021) A guide to understanding endoplasmic reticulum stress in metabolic disorders. Mol Metab 47:101169. https://doi.org/10.1016/j.molmet.2021.101169
Ren J, Bi Y, Sowers JR et al (2021) Endoplasmic reticulum stress and unfolded protein response in cardiovascular diseases. Nat Rev Cardiol 18:499–521. https://doi.org/10.1038/s41569-021-00511-w
Guo X, Shi Q, Zhang W et al (2022) Lipid droplet—a new target in ischemic heart disease. J Cardiovasc Transl Res 15:730–739. https://doi.org/10.1007/s12265-021-10204-x
Silva-Palacios A, Zazueta C, Pedraza-Chaverri J (2020) ER membranes associated with mitochondria: possible therapeutic targets in heart-associated diseases. Pharmacol Res 156:104758. https://doi.org/10.1016/j.phrs.2020.104758
Wu X, Liu Z, Yu XY et al (2021) Autophagy and cardiac diseases: therapeutic potential of natural products. Med Res Rev 41:314–341. https://doi.org/10.1002/med.21733
Pires Da Silva J, Monceaux K, Guilbert A et al (2020) SIRT1 protects the heart from ER stress-induced injury by promoting eEF2K/eEF2-dependent autophagy. Cells. https://doi.org/10.3390/cells9020426
Arrieta A, Blackwood EA, Stauffer WT, Glembotski CC (2019) Integrating ER and mitochondrial proteostasis in the healthy and diseased heart. Front Cardiovasc Med 6:193. https://doi.org/10.3389/fcvm.2019.00193
Li J, Zhang D, Wiersma M, Brundel B (2018) Role of autophagy in proteostasis: friend and foe in cardiac diseases. Cells. https://doi.org/10.3390/cells7120279
Glembotski CC (2014) Roles for ATF6 and the sarco/endoplasmic reticulum protein quality control system in the heart. J Mol Cell Cardiol 71:11–15. https://doi.org/10.1016/j.yjmcc.2013.09.018
Amen OM, Sarker SD, Ghildyal R, Arya A (2019) Endoplasmic reticulum stress activates unfolded protein response signaling and mediates inflammation, obesity, and cardiac dysfunction: therapeutic and molecular approach. Front Pharmacol 10:977. https://doi.org/10.3389/fphar.2019.00977
Kutschka I, Bertero E, Wasmus C et al (2023) Activation of the integrated stress response rewires cardiac metabolism in barth syndrome. Basic Res Cardiol 118:47. https://doi.org/10.1007/s00395-023-01017-x
Li J, Horak KM, Su H et al (2011) Enhancement of proteasomal function protects against cardiac proteinopathy and ischemia/reperfusion injury in mice. J Clin Invest 121:3689–3700. https://doi.org/10.1172/JCI45709
Willemsen N, Arigoni I, Studencka-Turski M et al (2022) Proteasome dysfunction disrupts adipogenesis and induces inflammation via ATF3. Mol Metab 62:101518. https://doi.org/10.1016/j.molmet.2022.101518
Steffen J, Seeger M, Koch A, Kruger E (2010) Proteasomal degradation is transcriptionally controlled by TCF11 via an ERAD-dependent feedback loop. Mol Cell 40:147–158. https://doi.org/10.1016/j.molcel.2010.09.012
Sha Z, Goldberg AL (2014) Proteasome-mediated processing of Nrf1 is essential for coordinate induction of all proteasome subunits and p97. Curr Biol 24:1573–1583. https://doi.org/10.1016/j.cub.2014.06.004
Tomlin FM, Gerling-Driessen UIM, Liu YC et al (2017) Inhibition of NGLY1 inactivates the transcription factor Nrf1 and potentiates proteasome inhibitor cytotoxicity. ACS Cent Sci 3:1143–1155. https://doi.org/10.1021/acscentsci.7b00224
Koizumi S, Irie T, Hirayama S et al (2016) The aspartyl protease DDI2 activates Nrf1 to compensate for proteasome dysfunction. Elife. https://doi.org/10.7554/eLife.18357
Bartelt A, Widenmaier SB, Schlein C et al (2018) Brown adipose tissue thermogenic adaptation requires Nrf1-mediated proteasomal activity. Nat Med 24:292–303. https://doi.org/10.1038/nm.4481
Widenmaier SB, Snyder NA, Nguyen TB et al (2017) NRF1 Is an ER membrane sensor that is central to cholesterol homeostasis. Cell 171:1094–1109. https://doi.org/10.1016/j.cell.2017.10.003
Cui M, Atmanli A, Morales MG et al (2021) Nrf1 promotes heart regeneration and repair by regulating proteostasis and redox balance. Nat Commun 12:5270. https://doi.org/10.1038/s41467-021-25653-w
Kotschi S, Jung A, Willemsen N et al (2022) NFE2L1-mediated proteasome function protects from ferroptosis. Mol Metab 57:101436. https://doi.org/10.1016/j.molmet.2022.101436
Predmore JM, Wang P, Davis F et al (2010) Ubiquitin proteasome dysfunction in human hypertrophic and dilated cardiomyopathies. Circulation 121:997–1004. https://doi.org/10.1161/CIRCULATIONAHA.109.904557
Makris N, Georgiopoulos G, Laina A et al (2023) Cardiac mechanics in response to proteasome inhibition: a prospective study. Eur Heart J Cardiovasc Imaging 24:643–652. https://doi.org/10.1093/ehjci/jeac168
Batista-Almeida D, Martins-Marques T, Ribeiro-Rodrigues T, Girao H (2020) The role of proteostasis in the regulation of cardiac intercellular communication. Adv Exp Med Biol 1233:279–302. https://doi.org/10.1007/978-3-030-38266-7_12
Chang TL, Chang CJ, Lee WY et al (2009) The roles of ubiquitin and 26S proteasome in human obesity. Metabolism 58:1643–1648. https://doi.org/10.1016/j.metabol.2009.05.020
Brody MJ, Vanhoutte D, Bakshi CV et al (2019) Disruption of valosin-containing protein activity causes cardiomyopathy and reveals pleiotropic functions in cardiac homeostasis. J Biol Chem 294:8918–8929. https://doi.org/10.1074/jbc.RA119.007585
Dominguez F, Cuenca S, Bilinska Z et al (2018) Dilated cardiomyopathy due to BLC2-associated athanogene 3 (BAG3) mutations. J Am Coll Cardiol 72:2471–2481. https://doi.org/10.1016/j.jacc.2018.08.2181
Alcalai R, Arad M, Wakimoto H et al (2021) LAMP2 cardiomyopathy: consequences of impaired autophagy in the heart. J Am Heart Assoc 10:e18829. https://doi.org/10.1161/JAHA.120.018829
Farini A, Gowran A, Bella P et al (2019) Fibrosis rescue improves cardiac function in dystrophin-deficient mice and duchenne patient-specific cardiomyocytes by immunoproteasome modulation. Am J Pathol 189:339–353. https://doi.org/10.1016/j.ajpath.2018.10.010
Kotter S, Andresen C, Kruger M (2014) Titin: central player of hypertrophic signaling and sarcomeric protein quality control. Biol Chem 395:1341–1352. https://doi.org/10.1515/hsz-2014-0178
Zech ATL, Prondzynski M, Singh SR et al (2022) ACTN2 mutant causes proteopathy in human iPSC-derived cardiomyocytes. Cells. https://doi.org/10.3390/cells11172745
Verdonschot JAJ, Vanhoutte EK, Claes GRF et al (2020) A mutation update for the FLNC gene in myopathies and cardiomyopathies. Hum Mutat 41:1091–1111. https://doi.org/10.1002/humu.24004
Kitajima Y, Yoshioka K, Suzuki N (2020) The ubiquitin-proteasome system in regulation of the skeletal muscle homeostasis and atrophy: from basic science to disorders. J Physiol Sci 70:40. https://doi.org/10.1186/s12576-020-00768-9
Bonam SR, Wang F, Muller S (2019) Lysosomes as a therapeutic target. Nat Rev Drug Discov 18:923–948. https://doi.org/10.1038/s41573-019-0036-1
Chen Y, Guo Z, Li S et al (2021) Spermidine affects cardiac function in heart failure mice by influencing the gut microbiota and cardiac galectin‑3. Front Cardiovasc Med 8:765591. https://doi.org/10.3389/fcvm.2021.765591
Lee BH, Lee MJ, Park S et al (2010) Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature 467:179–184. https://doi.org/10.1038/nature09299
Kittleson MM, Maurer MS, Ambardekar AV et al (2020) Cardiac amyloidosis: evolving diagnosis and management: a scientific statement from the American Heart Association. Circulation 142:e7–e22. https://doi.org/10.1161/CIR.0000000000000792
Park CS, Cha H, Kwon EJ et al (2012) The chemical chaperone 4‑phenylbutyric acid attenuates pressure-overload cardiac hypertrophy by alleviating endoplasmic reticulum stress. Biochem Biophys Res Commun 421:578–584. https://doi.org/10.1016/j.bbrc.2012.04.048
Zeng S, Wang H, Chen Z et al (2018) Effects of geranylgeranylacetone upon cardiovascular diseases. Cardiovasc Ther 36:e12331. https://doi.org/10.1111/1755-5922.12331
Rani S, Sreenivasaiah PK, Kim JO et al (2017) Tauroursodeoxycholic acid (TUDCA) attenuates pressure overload-induced cardiac remodeling by reducing endoplasmic reticulum stress. PLoS ONE 12:e176071. https://doi.org/10.1371/journal.pone.0176071
Fontana L, Partridge L, Longo VD (2010) Extending healthy life span—from yeast to humans. Science 328:321–326. https://doi.org/10.1126/science.1172539
He C, Bassik MC, Moresi V et al (2012) Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature 481:511–515. https://doi.org/10.1038/nature10758
Acknowledgements
We thank all lab members for discussions and the enjoyable atmosphere. A.B. was supported by DFG (SFB1123-B10 and SPP2306 BA4925/2-1), the Deutsches Zentrum für Herz-Kreislauf-Forschung (DZHK), and the European Research Training Group (ERC) Starting Grant PROTEOFIT. We acknowledge BioRender.com for help with the figures. We apologize to colleagues whose work we could not cite due to space limitations.
Author information
Authors and Affiliations
Contributions
J.G. and L.M. wrote the manuscript and designed the figure A.B. read and commented on the figure and manuscript.
Corresponding author
Ethics declarations
Conflict of interest
J. Guerra, L. Matta and A. Bartelt declare that they have no competing interests.
For this article no studies with human participants or animals were performed by any of the authors. All studies mentioned were in accordance with the ethical standards indicated in each case.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
J. Guerra and L. Matta contributed equally to the manuscript.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Guerra, J., Matta, L. & Bartelt, A. Cardiac proteostasis in obesity and cardiovascular disease. Herz 49, 118–123 (2024). https://doi.org/10.1007/s00059-024-05233-6
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00059-024-05233-6