
Abstract
Hereditary spastic paraparesis (HSP) is a group of central nervous system diseases primarily affecting the spinal upper motor neurons, with different inheritance patterns and phenotypes. SPG46 is a rare, early-onset and autosomal recessive HSP, linked to biallelic GBA2 mutations. About thirty families have been described worldwide, with different phenotypes like complicated HSP, recessive cerebellar ataxia or Marinesco-Sjögren Syndrome. Herein, we report five SPG46 patients harbouring five novel GBA2 mutations, the largest series described in Italy so far. Probands were enrolled in five different centres and underwent neurological examination, clinical cognitive assessment, column imaging for scoliosis assessment, ophthalmologic examination, brain imaging, GBA2 activity in peripheral blood cells and genetic testing. Their phenotype was consistent with HSP, with notable features like upper gaze palsy and movement disorders. We review demographic, genetic, biochemical and clinical information from all documented cases in the existing literature, focusing on the global distribution of cases, the features of the syndrome, its variable presentation, new potential identifying features and the significance of measuring GBA2 enzyme activity.
Similar content being viewed by others


Introduction
Hereditary spastic paraparesis (HSP) represents a group of central nervous system (CNS) diseases that mainly involve the spinal portion of upper motor neurons [1]. Hallmark pathologic alteration in HSPs is diffuse axonal “dying-back” degeneration, most represented in the terminal segments of the longest axons, with possible involvement of dorsal columns [2, 3]. Involvement of the lower motor neurons can also be observed [4]. HSPs have an estimated prevalence ranging from 2 to 4.1 in 104 individuals [5, 6], and they are classified according to inheritance (autosomal dominant, autosomal recessive, X-linked or mitochondrial with maternal trait transmission), to phenotype (“pure” or “complex”) and to onset (early or late) [7, 8]. The pathogenesis of HSP is connected to a wide range of cellular processes, including membrane and axonal transport, modulation of the endoplasmic reticulum membrane, mitochondrial function, DNA repair, autophagy, lipid metabolism and myelination. Additionally, dysfunction in endosome membrane trafficking, oxidative stress and mitochondrial DNA polymorphisms have been implicated [9].
Phenotypically, “pure” HSPs [10] typically manifest with pyramidal signs starting in lower limbs, associated with variable disorders as sphincter dysfunctions and deep sensory loss. On the other hand, “complex” HSPs may display a broader range of neurological manifestations including cerebellar dysfunction, peripheral neuropathy, extrapyramidal features, seizures, deafness, cognitive impairment and psychiatric disorders [1, 5]. Extraneurological manifestations can include cataracts, optic neuropathy, retinitis pigmentosa, facial dysmorphisms, scoliosis, hip dislocation and different foot deformities [1, 11]. Furthermore, CNS neuroimaging reveals characteristic features, often related to a specific subtype: cerebellar atrophy, thin corpus callosum (TCC), white matter abnormalities (WMA), spinal cord atrophy, brain iron accumulation and hydrocephalus [12,13,14]. Within the group of autosomal recessive hereditary spastic paraplegias (ARHSP), SPG11 is the most common, followed by SPG7, SPG15 and SPG56, whereas SPG46 seems more rare [15,16,17,18,19,20,21].
SPG46 is a rare, early onset spastic paraparesis, classified as “complex” [5]. It is inherited in an autosomal recessive manner, and its clinical presentation has appeared strikingly different from other ARHSP since its initial and seminal description [22]. It is associated with biallelic mutations in the GBA2 gene (locus 9p13.3) encoding for the non-lysosomal glucosylceramidase (GBA2) [20, 23], a ubiquitous enzyme associated with the endoplasmic reticulum and the plasma membrane, which catalyzes conversion of glucosylceramide to glucose and ceramide [24]. The other (non-homologous) enzyme degrading glucosylceramide is the lysosomal acid β-glucosidase (GBA), whose mutations cause Gaucher’s disease and a form of hereditary Parkinson’s disease [25]. Glucosylceramide is the precursor component of gangliosides. Mutations in the GBA2 lead to changes in enzymatic activity, which can be detected in lymphoblasts and leucocytes of affected subjects [26], but also affect neurons [23, 27], resulting in abnormal increase of glucosylceramide, although the pathogenic mechanism of neurodegeneration is still unclear [24]. GBA2 mutations seem to lead to a clinical spectrum that encompasses different phenotypes, including HSP, autosomal recessive cerebellar ataxia (ARCA), or a more complex and severe condition like Marinesco-Sjögren Syndrome (MSS) [18, 27, 28]. Clinically, HSP46 may show plethoric signs, both neurological and extraneurological, besides spastic paraparesis, such as: cerebellar dysfunction, peripheral neuropathy, distal amyotrophy, cognitive impairment, scoliosis, cataracts [16, 20, 29], and there is still not a consensus basis about the overall definite phenotype. Furthermore, not all reports contain information about the features thought to be hallmarks, or at least common, in this disease (Table 1). Brain MRI may display WMA, TCC, cerebral, brainstem and cerebellar atrophy. To date, more than 90 genetic types of HSP have been identified [5, 15] (https://neuromuscular.wustl.edu/spinal/fsp.html; OMIM), and 62 patients, between isolated cases and families, with SPG46 have been described worldwide. Since its clinical discovery [22], when its genetic mutation was still unknown, and subsequent identification of the causative mutation [16], 23 reports about SPG46 have been published. In over 15 years, different phenotypes have been described, pointing out a phenotypical heterogeneity and reinforcing the concept of “clinical spectrum” of this disease. Herein we report five novel GBA2 pathogenic variants detected in unrelated SPG46 Italian patients. We also provide a comprehensive review of demographic, genetic, biochemical and clinical data from all SPG46 cases described in the existing literature, discussing about cases’ global distribution, overall phenotype’s characteristics, variable expressivity, possible hallmarks and the importance of GBA2 activity dosage.
Methods
Patients
This multicentric case series study was performed in accordance with the Declaration of Helsinki and its later amendments. Written informed consent and ethical approval (CE Lazio) were obtained. In a single laboratory, we tested about 735 patients with clinical evidence of HSP without genetic diagnosis, using a multigene targeted resequencing panel and investigating the coding exons and flanking introns of the genes known to be associated with HSPs [5, 15, 30]. From September to November 2021, five patients harbouring biallelic GBA2 mutations (four men; one woman) were identified and recruited in five Italian neurology centres (University of Rome Sapienza, Policlinico Universitario A. Gemelli IRCCS in Rome, “Città della Salute e della Scienza” in Torino, IRCCS Stella Maris Foundation in Pisa, University of Messina). These patients were enrolled in the study and underwent further investigation and analysis. Family and clinical history were collected. All patients underwent neurological examination, clinical cognitive assessment through Mini Mental State Examination (MMSE) [31], spinal radiological study (for scoliosis assessment), ophthalmologic examination (for cataract assessment), brain MRI (Table 1).
GBA2 activity measurement
Two of five index cases (Table 1; Fig. 1, proband A and B) underwent GBA2 enzyme dosage [32]. Leucocytes were isolated from ~ 6-mL EDTA blood as previously described [33], and kept at − 20 °C for 1 day before being thawed and maintained on ice until the start of the enzyme reaction. For the isolation of leucocytes, 10% dextran was added. Erythrocytes were allowed to sediment at room temperature for 45 min and the upper phase centrifuged at 1125 × g for 10 min. The pellet was washed with 0.9% NaCl and stored at − 20 °C until the analysis. Protein content was determined by the BCA protein assay (Pierce, Rockford, USA). Leucocytes were diluted in 100 µL of deionized water and sonicated three times for 2 s by a Vibra Cell Sonicator (Sonics-Materials Inc., VCX130, Newton, Connecticut, USA). Stock solutions of substrate and inhibitor were prepared as follows: Substrate: 4-methylumbelliferone-β-glucopyranoside 10 mM (Glycosinth, Warrington, UK) in citric acid 200 mM (Sigma-Aldrich, St. Louis, MO, USA) and disodium hydrogen phosphate 100 mM (Merck, VWR International), pH 5.2. Inhibitor: conduritol β epoxide (CBE) (0.26 M) (Sigma-Aldrich, St. Louis, MO, USA)). Ten microliters of sample (30-µg protein) with and without 10 µL of inhibitor were made up to 50 µl with deionized water and pre-incubated for 1 min at 37 °C. The assay was initiated by adding 100 µl of substrate. Duplicates of each sample with and without inhibitor, as well as duplicates blanks (10 µl of deionized water and 100 µl of substrate) were incubated in 2-mL eppendorf tubes for 120 min at 37 °C in a water bath with orbital shaker. The reaction was stopped with (0.5 M) NaHCO3/(0.5 M) Na2CO3 pH 10.3 and fluorescence of samples, blanks and standard solution (4-methyl umbelliferone 0.5 mM, diluted in (0.5 M) NaHCO3/(0.5 M) Na2CO3 pH 10.3) were measured within 1 h (shielded from light) in a spectrofluorometer ( FL6500 Perkin Elmer, Waltham, MA, USA) using an excitation wavelength of 365 nm and an emission wavelength of 448 nm. The activity of GBA2 was calculated according to the standard curve and expressed as nmol/mg proteins.

Families’ pedigree; family’s letter indicates each proband; arrows indicate the probands; chromatograms and mutations on bottom of each pedigree
Molecular and database search
DNA extraction was carried out using peripheral blood lymphocytes obtained from the patients. Subsequently, Next Generation Sequencing (NGS) analysis was performed using an amplicon based customized NGS panel (Illumina TrueSeq Custom Amplicon, TSCA) including more than 200 genes involved in HSP pathogenesis. Literature was reviewed using PubMed and Google Scholar, and findings were collected in Tables 3, 4 and 5. Excel was employed to create map graphics. Search for variants of GBA2 was done using population databases (dbSNP, 1000genome, EVS) and local databases, and their pathogenicity were assessed according to the American College of Medical Genetics and Genomics (ACMG) guidelines [34].
Results
Cases clinical reports
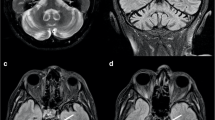
Patients’ clinical, imaging and laboratory features are summarized in Table 1. Pedigrees are shown in Fig. 1. Consanguinity of the patients’ parents was identified in two out of five cases (Fig. 1, families A and E). All families, except for one, had Italian descent (Family A came from Morocco—Table 2, Figs.1 and 2). Except for one proband, all individuals had early disease onset (between 6 and 7 years—mean 6.8 years). In one case (proband B) we found congenital onset. The overall initial manifestation was spasticity in the lower limbs (5/5), subsequently followed by various neurological and extraneurological manifestations which, with some degree of variability, enriched the clinical picture (Table 1): cerebellar syndrome (4/5), sphincteric symptoms (like urge incontinence, 3/5), intellectual disability (MCI, 4/5), peripheral neuropathy (4/5), movement disorders like UL tremor and dystonia (1/5), bilateral cataracts (4/5), scoliosis (2/5) and pes cavus (1/5). Mean SPRS score was 23.75 (NA in one case). At the time of last neurological examination, the mean age of the patients was 38.6 years. In two patients, we found ocular movement disorder (upper gaze palsy (UGP)). MMSE assessment was administered, mainly resulting in MCI (except proband A). No one of the male patients had hypogonadism. They all underwent spine RX and brain MRI: the most frequent sign was WMA (4/5), followed by TCC (2/5). Two patients had skeletal deformities, comprising scoliosis and/or pes cavus. Disease course was slowly progressive (mean 32 years at the time of last examination).

A Families’ distribution per country; the colour’s intensity shows the prevalence. B Cases’ distribution per country; the colour’s intensity shows the prevalence
Molecular findings
Gene testing identified 7 GBA2 variants, four of which were compound heterozygous (Fig. 1). The c.472G > A (p.Gly158Arg) and c.2063 G > A (p.Cys688Thr) pathogenic variants have already been reported [35, 36]. The other five variants, i.e. the homozygous c.1786 G > T (p.Gly596Trp) and the four compound heterozygous (c.1846_1860delinsTCAGTCCCGATA + c.2020C > T (p.Gln674*) and c.1653G > A (p.Trp551*) + c.1322C > A (p.Ala441Glu), were absent in our in-house databases as well as in population databases (dbSNP, 1000genome, gnomAD) and were classified as likely pathogenic according to the ACMG guidelines. Missense variants were indicated as “probably damaging” and “damaging” by two in silico predictors (PolyPhen-2 and SIFT).
Biochemical findings
Two patients underwent analysis of enzymatic activity, while it was not available for three. We determined the GBA2 activity in leucocytes as the beta-glucosidase activity that is resistant to 2.5 mM conduritol B epoxide. This activity was much reduced in proband A and B (Table 1; 0.28 nmol/mg and 0 0.01 nmol/mg, respectively) by comparison with the mean values of 5 controls (3.9; reference range: 2.5 to 5.3 nmol/mg). We acknowledge that this method of measuring GBA2 activity may lead to underestimating GBA2 activity [37].
Discussion
We present five previously unreported Italian patients with SPG46. These patients were found to have five novel GBA2 variants, all of which were determined to be pathogenic based on in silico predictors. This report represents the 24th documented study on the disease (Table 3, 4 and 5). Thus far, a total of 67 cases (30 men, 34 women, sex not specified in three) from 36 families have been described worldwide (Tables 2 and 3; Fig. 2) [16,17,18,19,20,21, 27, 29, 35, 36, 38,39,40,41,42,43,44,45,46,47,48,49,50] since the seminal description of Boukhris et al. in 2008 [22]. Patients with GBA2 pathogenic variants have been described in Tunisia, Belgium, Turkey, Portugal, Cyprus, Italy, Romania, Netherlands, China, Norway, France, Saudi Arabia, Japan, Germany, India, Taiwan, USA and Spain [16,17,18,19,20,21, 27, 29, 35, 36, 38,39,40,41,42,43,44,45,46,47,48,49,50] (Fig. 2 and Table 2). Based on the worldwide distribution, the prevalence of the disease seems to be higher in the Mediterranean area (Fig. 2). It ought to be considered the prevalence in each country where the highest number of cases has been reported (Table 2). In Tunisia, for instance, there have been 15 cases reported, with a prevalence of approximately 15 in 1.2 × 107 individuals. Similarly, in Italy, there have been 10 reported cases in approximately 5.9 × 107 individuals. Saudi Arabia has reported 8 cases, resulting in a prevalence of about 8 in 3.6 × 107 individuals. Lastly, in China, 4 cases have been documented, indicating a prevalence of around 4 in 140 × 107 individuals. Considering these prevalence rates, the Mediterranean area exhibits the highest concentration of SPG46 cases due to its smaller population (Fig. 2). For instance, this is particularly notable when comparing it with countries like the USA, which has a significantly larger population (approximately 33 × 107 inhabitants) but a minimal prevalence of the disease, with only one reported case so far [35]. Since ARHSPs are more common in countries with a higher rate of consanguinity, this may provide an explanation for the higher number of reported cases in these areas. A thorough demographical, clinical, radiological and biochemical comparison was conducted, examining the features of our cases in relationship with the available literature (Tables 1, 3, 4 and 5). Two out of five patients included in this study belonged to consanguineous families, where the parents were found to be relatives (Fig. 1; Table 1). Consanguinity is commonly observed in recessive diseases, and SPG46 makes no exception. Among the available reports in literature with this information (12 out of 23), consanguinity was investigated in 19 families (out of 31) and was found in 14 of them (73.68%) (Table 3). This highlights the prevalence of consanguineous marriages within the context of SPG46 and underscores the significance of genetic factors in the disease’s inheritance patterns. In all cases reported so far, the presence of both spastic paraparesis and cerebellar syndrome has been consistently observed (Table 3). In our study, all patients had early onset (6.8 year) and slow progression over time. Remarkably, one case had congenital onset (Table 1; proband B), and one had the longest progression so far (Table 1; proband C—44 years of disease); it is the second reported with such disease duration [47, 50]. Additional clinical features, commonly regarded as characteristic signs of this rare HSP [16,17,18,19,20, 22] such as neuropathy, MCI, bilateral cataracts, scoliosis, pes cavus and hypogonadism are observed with varying prevalence among the SPG46 population (Table 3). MCI is a common feature (Tables 1 and 3), but it may show very lately [18]. About half of the cases described so far show MCI, but its prevalence may turn out to be higher, due to later onset, as in our proband C (Table 1).
Movement disorders, like head and upper limbs’ tremor, cranial and upper limbs’ dystonia, can be observed with moderate occurrence, and they appear to be part of the clinical presentation in several described cases (17 out of 55—Table 4). Cervical dystonia has been outlined as the onset symptom in one patient, later evolved into a complex athetotic-dystonic disorder which involved the UL (initially described as “writer’s cramp”); at brain MRI she showed brainstem atrophy, also involving basal ganglia [47]. Facial myokymias were reported too [29]. Other neurological signs and symptoms emerged (Tables 1 and 4). Six cases of hearing loss have been reported. This is a symptom frequently found in mitochondrial diseases [51, 52]. Since a role in mitochondrial fragmentation has been already outlined in GBA2 mutation [53], we may suppose a similar mechanism in SPG46. Four cases of psychiatric disorders are also described [36, 43, 46]: in one case, the disease onset was represented by delusions [43]. Among the other neurological signs, UGP is the most frequent (19%—Tables 1 and 4) [18, 21, 47, 48]. Interestingly, this phenomenon is frequently observed in Gaucher’s Disease Type 3 (GD3). It is attributed to ceramide accumulation in cerebellar and brainstem areas controlling vertical gaze: floccular lobe, vestibular system, pontine paramedian reticular formation, rostral interstitial nucleus of the medial longitudinal fascicle and motor neurons of the abducens nucleus [54, 55]. GD3 is a neurodegenerative disease caused by pathological accumulation of glucosylceramide in the CNS due to lysosomal GBA dysfunction [56], similarly to what happens in SPG46. GBA and GBA2 do not have the same location or functioning [57]. However, it has been pointed out not only an akin role (i.e. glucosylceramide metabolism), but also an indirect action synergy [58]. Malekkou et al. biochemically characterized the same Cypriot SPG46 family described in 2014 [18], and, besides abolished GBA2 activity, they highlighted a compensatory effect of GBA, since its activity was threefold higher in SPG46 patients compared to controls [26]. Thus, the two enzymes not only share a similar role, but seem also to be related. Since UGP seems to be recurrent in SPG46 (Tables 1 and 4), we hypothesize a similar role of GBA2, resulting in brainstem and cerebellar dysfunction, and thus leading to UGP.
With exception of peripheral neuropathy and cognitive assessment, many reports lack additional clinical data or do not provide negative results (Tables 3, 4 and 5). As shown in Table 3, features such as scoliosis, foot abnormalities and hypogonadism should undergo more comprehensive investigation to determine their status as defining characteristics. This presents a challenge in further delineating the phenotypic profile of SPG46. Furthermore, manifestations like movement disorders (dystonia), ocular movements abnormalities and skeletal deformities might have a higher occurrence (Table 4). In future reports, recognizing and considering these features can aid diagnosis.
In most reports (20 out of 24, which includes our own study—Table 5) patients and families are primarily described with a phenotype consistent with HSP. Only few descriptions classify the disease as ARCA [17,18,19], and just one report identifies it as MSS [27]. That may often depend on predominant symptoms, or on signs and symptoms at onset. In 2013, Hammer et al. discovered the second group of GBA2-mutated patients, and initially diagnosed the condition as autosomal recessive ataxia, as the first presentation involved cerebellar syndrome. However, shortly thereafter, in addition to peripheral neuropathy, significant spasticity emerged, initially in the lower limbs and subsequently extending to the upper limbs, becoming highly pronounced and dominating the overall clinical presentation [19]. Later, Votsi et al. discovered a new GBA2-mutated family, in Cyprus, and classified it as ARCA. However, their phenotype description involves a typical HSP onset and progression (with spasticity in lower limbs) [18]. The first Italian description of SPG46, depicted as ARCA, involved three affected individuals from the same family. Still, a predominantly cerebellar phenotype was evident in a single case, while the other members displayed HSP, and significant intrafamilial variability [17]. Curiously, the variant discovered by Hammer et al. (c.2618G > A) [19] is the same of the Saudi SPG46 family from 2019 [44], which was described mainly as a HSP, complicated by cerebellar ataxia. Thus, different phenotypes may arise from same identical mutations, as also evidenced by the intrafamilial variability observed by Citterio et al. [17]. There are cases where the same genotype causes different degrees of phenotypical expression, even in the same family, as seen in diseases like neurofibromatosis, Van der Woude syndrome or holoprosencephaly [59,60,61]. Differently from incomplete penetrance, in which the expected phenotype manifests or not, this phenomenon is referred to as “variable expressivity”, which quantifies the degree to which a genotype displays its phenotypic expression [62]. Variable expressivity appears to be caused by a range of factors, including common variants, variants in regulatory regions, gene-modifiers, epigenetics, environmental factors and lifestyle [63]. In the two Norwegian families described in 2017, the probands presented at examination with early onset cerebellar ataxia, also with bilateral cataract, mental retardation and late spastic paraparesis [27]. Clinical diagnosis of MSS was made (one of the families was visited and diagnosed in 1977) [64]. MSS is an AR disorder, caused by mutations in SIL1, characterized by cerebellar atrophy with ataxia, early-onset cataracts, and it also may include mild to severe intellectual disability, hypogonadism and skeletal abnormalities [65,66,67]. Its clinical hallmarks are child-onset hypotonia and muscle weakness but not spasticity in lower limbs [68]. The Norwegian patients did not exhibit hypotonia or myopathy during childhood and instead presented with early onset-spastic paraparesis. Therefore, SPG46 may start with diverse symptoms sometimes different from spasticity in lower limbs, like cerebellar ataxia and may seldom show different phenotypes [17,18,19, 27]. Regardless of different onset or dominant symptoms, the disease is primarily described in most reports as a complex form of ARHSP. Considering the long disease duration (mean 26.9 years—Tables 1 and 3), the overall phenotype gradually manifests as a “typical” complex ARHSP over time, exhibiting distinct features that serve as hallmarks of SPG46 (Tables 1, 3, 4 and 5). At times, the differentiation between cerebellar ataxia and HSP seems artificial, as it reveals a phenotypic continuum linked to specific genes, which can be better understood through the concept of variable expressivity: indeed, little is known about any cis–trans regulatory elements of GBA2.
Our MRI findings demonstrate slight variations compared to those reported in the literature (Tables 1 and 5). Specifically, we observed a lower incidence of cerebellar atrophy (0/5), a higher incidence of brainstem atrophy (40% versus 7%), and significant differences concerning WMA (80% versus 15%). These findings highlight the importance of WMA sign in our cohort. However, it is worth noting that our study includes a limited number of cases, and further expansion of the cohort is necessary to enable a more meaningful comparison (Tables 1 and 5).
We have noted a significant reduction in GBA2 activity, as confirmed by tests conducted in our study as well in a few others [20, 23, 26, 27, 29, 47]. Importantly, previous reports and mice studies suggest that this enzyme, despite being relatively understudied, may affect axonal differentiation and branching [69] and locomotor function [20, 24]. Also, GBA2 shows species-specificity [70, 71], especially regarding male reproduction [72, 73], implying that there is more than meets the eye. GBA2 activity test is undoubtedly useful from a diagnostic point of view (Table 1). In our congenital case, it is noteworthy that GBA2 activity was nearly absent, exhibiting a significant reduction compared to proband A, who presented a more typical onset and progression (Table 1).
Conclusion
In this study, we present a large cohort of Italian SPG46 patients, harbouring a total of seven GBA2 variants, five of which are novel. The associated phenotype aligns with the overall SPG46 phenotype described in previous reports, showing characteristic features of “typical” ARHSP syndrome and hallmarks like early onset and slow progression of disease, cognitive impairment, scoliosis and cataract. Noteworthy, one case represents the first documented congenital presentation of this condition, and another has the longest disease duration thus far. We identified UGP in two cases of our cohort. It also appears to be relatively frequent in the existing literature. Additionally, movement disorders like tremor and dystonia were observed in several patients. We propose a possible underlying mechanism for UGP and point out the clinical value of this symptom. Also, we highlight tremor and dystonic disorders as clinical specific traits. Unfortunately, there is a high number of papers with unreported data (Tables 3, 4 and 5), limiting informativeness for natural history studies. Brain MRI displayed distinctive SPG46 features, with higher WMA incidence. Dosage of GBA2 activity can be highly beneficial in the diagnostic process, to reduce the time to a confirmatory gene testing. The minor GBA2 activity in our congenital case may suggest a correlation between enzymatic activity and onset, duration, or severity of the disease; but it is hard to speculate on the possible correlations between clinical features and residual enzyme activity since the number of enzyme tests is poor. Biochemical tests can aid in early diagnosis, interdisciplinary management, and assist in potential therapeutic interventions. Future availability of glucosylceramide synthase inhibitors [74, 75] may constitute an efficient and cost-effective genetic testing strategy. It is vital to emphasize the significance of comprehensive observational reports, as they provide valuable information, which enhance our understanding of ultra-rare diseases such as SPG46.
Data Availability
The authors confirm that the data supporting the findings of this study are available within the article (specifically, Tables 1, 2, 3, 4, 5 and Figures 1, 2A, 2B). More specific clarifications about data are available, upon reasonable request, from the corresponding author (E.C.).
References
Salinas S, Proukakis C, Crosby A, Warner TT (2008) Hereditary spastic paraplegia: clinical features and pathogenetic mechanisms. Lancet Neurol 7:1127–1138. https://doi.org/10.1016/S1474-4422(08)70258-8
Behan WMH, Maia M (1974) Strumpell’s familial spastic paraplegia: genetics and neuropathology. J Neurol Neurosurg Psychiatry 37:8–20. https://doi.org/10.1136/jnnp.37.1.8
DeLuca GC, Ebers GC, Esiri MM (2004) The extent of axonal loss in the long tracts in hereditary spastic paraplegia: axonal loss in hereditary spastic paraplegia. Neuropathol Appl Neurobiol 30:576–584. https://doi.org/10.1111/j.1365-2990.2004.00587.x
Parodi L, Fenu S, Stevanin G, Durr A (2017) Hereditary spastic paraplegia: more than an upper motor neuron disease. Rev Neurol 173:352–360. https://doi.org/10.1016/j.neurol.2017.03.034
Lallemant-Dudek P, Durr A (2021) Clinical and genetic update of hereditary spastic paraparesis. Rev Neurol 177:550–556. https://doi.org/10.1016/j.neurol.2020.07.001
Ruano L, Melo C, Silva MC, Coutinho P (2014) The global epidemiology of hereditary ataxia and spastic paraplegia: a systematic review of prevalence studies. Neuroepidemiology 42:174–183. https://doi.org/10.1159/000358801
Finsterer J, Löscher W, Quasthoff S et al (2012) Hereditary spastic paraplegias with autosomal dominant, recessive, X-linked, or maternal trait of inheritance. J Neurol Sci 318:1–18. https://doi.org/10.1016/j.jns.2012.03.025
de Souza PVS, de Rezende Pinto WBV, de RezendeBatistella GN et al (2017) Hereditary spastic paraplegia: clinical and genetic hallmarks. Cerebellum 16:525–551. https://doi.org/10.1007/s12311-016-0803-z
Lo Giudice T, Lombardi F, Santorelli FM et al (2014) Hereditary spastic paraplegia: clinical-genetic characteristics and evolving molecular mechanisms. Exp Neurol 261:518–539. https://doi.org/10.1016/j.expneurol.2014.06.011
Harding AE (1981) Hereditary “pure” spastic paraplegia: a clinical and genetic study of 22 families. J Neurol Neurosurg Psychiatry 44:871–883. https://doi.org/10.1136/jnnp.44.10.871
Hedera P. Hereditary Spastic Paraplegia Overview. 2000 Aug 15 [Updated 2021 Feb 11]. In: Adam MP, Feldman J, Mirzaa GM et al (eds) GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1509/
Agosta F, Scarlato M, Spinelli EG et al (2015) Hereditary spastic paraplegia: beyond clinical phenotypes toward a unified pattern of central nervous system damage. Radiology 276:207–218. https://doi.org/10.1148/radiol.14141715
Hourani R, El-Hajj T, Barada WH et al (2009) MR imaging findings in autosomal recessive hereditary spastic paraplegia. AJNR Am J Neuroradiol 30:936–940. https://doi.org/10.3174/ajnr.A1483
da Graça FF, de Rezende TJR, Vasconcellos LFR et al (2018) Neuroimaging in hereditary spastic paraplegias: current use and future perspectives. Front Neurol 9:1117. https://doi.org/10.3389/fneur.2018.01117
Klebe S, Stevanin G, Depienne C (2015) Clinical and genetic heterogeneity in hereditary spastic paraplegias: from SPG1 to SPG72 and still counting. Rev Neurol 171:505–530. https://doi.org/10.1016/j.neurol.2015.02.017
Boukhris A, Feki I, Elleuch N et al (2010) A new locus (SPG46) maps to 9p21.2-q21.12 in a Tunisian family with a complicated autosomal recessive hereditary spastic paraplegia with mental impairment and thin corpus callosum. Neurogenetics 11:441–448. https://doi.org/10.1007/s10048-010-0249-2
Citterio A, Arnoldi A, Panzeri E et al (2014) Mutations in CYP2U1, DDHD2 and GBA2 genes are rare causes of complicated forms of hereditary spastic paraparesis. J Neurol 261:373–381. https://doi.org/10.1007/s00415-013-7206-6
Votsi C, Zamba-Papanicolaou E, Middleton LT et al (2014) A novel GBA2 gene missense mutation in spastic ataxia. Ann Hum Genet 78:13–22. https://doi.org/10.1111/ahg.12045
Hammer MB, Eleuch-Fayache G, Schottlaender LV et al (2013) Mutations in GBA2 cause autosomal-recessive cerebellar ataxia with spasticity. Am J Hum Genet 92:245–251. https://doi.org/10.1016/j.ajhg.2012.12.012
Martin E, Schüle R, Smets K et al (2013) Loss of function of glucocerebrosidase GBA2 is responsible for motor neuron defects in hereditary spastic paraplegia. Am J Hum Genet 92:238–244. https://doi.org/10.1016/j.ajhg.2012.11.021
Yang Y-J, Zhou Z-F, Liao X-X et al (2016) SPG46 and SPG56 are rare causes of hereditary spastic paraplegia in China. J Neurol 263:2136–2138. https://doi.org/10.1007/s00415-016-8256-3
Boukhris A, Stevanin G, Feki I et al (2008) Hereditary spastic paraplegia with mental impairment and thin corpus callosum in Tunisia: SPG11, SPG15, and further genetic heterogeneity. Arch Neurol 65:393–402. https://doi.org/10.1001/archneur.65.3.393
Harzer K, Yildiz Y, Beck-Wödl S (2019) Assay of β-glucosidase 2 (GBA2) activity using lithocholic acid β-3-O-glucoside substrate for cultured fibroblasts and glucosylceramide for brain tissue. Biol Chem 400:745–752. https://doi.org/10.1515/hsz-2018-0438
Woeste MA, Wachten D (2017) The enigmatic role of GBA2 in controlling locomotor function. Front Mol Neurosci 10:386. https://doi.org/10.3389/fnmol.2017.00386
Riboldi GM, Di Fonzo AB (2019) GBA, Gaucher disease, and Parkinson’s disease: from genetic to clinic to new therapeutic approaches. Cells 8:364. https://doi.org/10.3390/cells8040364
Malekkou A, Samarani M, Drousiotou A et al (2018) Biochemical characterization of the GBA2 c.1780G>C missense mutation in lymphoblastoid cells from patients with spastic ataxia. Int J Mol Sci 19:3099. https://doi.org/10.3390/ijms19103099
Haugarvoll K, Johansson S, Rodriguez CE et al (2017) GBA2 mutations cause a Marinesco-Sjögren-like syndrome: genetic and biochemical studies. PLoS One 12:e0169309. https://doi.org/10.1371/journal.pone.0169309
Sultana S, Reichbauer J, Schüle R et al (2015) Lack of enzyme activity in GBA2 mutants associated with hereditary spastic paraplegia/cerebellar ataxia (SPG46). Biochem Biophys Res Commun 465:35–40. https://doi.org/10.1016/j.bbrc.2015.07.112
Gatti M, Magri S, Di Bella D et al (2021) Spastic paraplegia type 46: novel and recurrent GBA2 gene variants in a compound heterozygous Italian patient with spastic ataxia phenotype. Neurol Sci 42:4741–4745. https://doi.org/10.1007/s10072-021-05463-0
D’Amore A, Tessa A, Casali C et al (2018) Next generation molecular diagnosis of hereditary spastic paraplegias: an Italian cross-sectional study. Front Neurol 9:981. https://doi.org/10.3389/fneur.2018.00981
Folstein MF, Folstein SE, McHugh PR (1975) “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 12:189–198. https://doi.org/10.1016/0022-3956(75)90026-6
Svennerholm L, Håkansson G, Månsson JE, Vanier MT (1979) The assay of sphingolipid hydrolases in white blood cells with labelled natural substrates. Clin Chim Acta 92:53–64. https://doi.org/10.1016/0009-8981(79)90396-6
Vespasiani-Gentilucci U, D’Amico J, De Vincentis A et al (2017) Platelet count may impact on lysosomal acid lipase activity determination in dried blood spot. Clin Biochem 50:726–728. https://doi.org/10.1016/j.clinbiochem.2017.02.013
Richards S, Aziz N, Bale S et al (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17:405–424. https://doi.org/10.1038/gim.2015.30
Gill S, Gill S, Williamson M, Avila JD (2023) A rare case of hereditary spastic paraplegia 46 with novel compound heterozygous GBA2 gene variants (P3–8.013). In: Sunday, April 23. Lippincott Williams & Wilkins, pp 3171
Spagnoli C, Schiavoni S, Rizzi S et al (2020) New biallelic GBA2 variant in a patient with SPG46. Clin Neurol Neurosurg 191:105676. https://doi.org/10.1016/j.clineuro.2020.105676
Ridley CM, Thur KE, Shanahan J et al (2013) β-Glucosidase 2 (GBA2) activity and imino sugar pharmacology. J Biol Chem 288:26052–26066. https://doi.org/10.1074/jbc.M113.463562
Kancheva D, Atkinson D, De Rijk P et al (2016) Novel mutations in genes causing hereditary spastic paraplegia and Charcot-Marie-Tooth neuropathy identified by an optimized protocol for homozygosity mapping based on whole-exome sequencing. Genet Med 18:600–607. https://doi.org/10.1038/gim.2015.139
van de Warrenburg BP, Schouten MI, de Bot ST et al (2016) Clinical exome sequencing for cerebellar ataxia and spastic paraplegia uncovers novel gene-disease associations and unanticipated rare disorders. Eur J Hum Genet 24:1460–1466. https://doi.org/10.1038/ejhg.2016.42
Morais S, Raymond L, Mairey M et al (2017) Massive sequencing of 70 genes reveals a myriad of missing genes or mechanisms to be uncovered in hereditary spastic paraplegias. Eur J Hum Genet 25:1217–1228. https://doi.org/10.1038/ejhg.2017.124
Coutelier M, Hammer MB, Stevanin G et al (2018) Efficacy of exome-targeted capture sequencing to detect mutations in known cerebellar ataxia genes. JAMA Neurol 75:591–599. https://doi.org/10.1001/jamaneurol.2017.5121
Coarelli G, Romano S, Travaglini L et al (2018) Novel homozygous GBA2 mutation in a patient with complicated spastic paraplegia. Clin Neurol Neurosurg 168:60–63. https://doi.org/10.1016/j.clineuro.2018.02.042
Wei Q, Dong H-L, Pan L-Y et al (2019) Clinical features and genetic spectrum in Chinese patients with recessive hereditary spastic paraplegia. Transl Neurodegener 8:19. https://doi.org/10.1186/s40035-019-0157-9
Algahtani H, Shirah B, Ullah I et al (2021) Autosomal recessive cerebellar ataxia with spasticity due to a rare mutation in GBA2 gene in a large consanguineous Saudi family. Genes Dis 8:110–114. https://doi.org/10.1016/j.gendis.2019.07.009
Guan R-Y, Wu J-J, Ding Z-T et al (2020) Clinical and genetic findings in a cohort of Chinese patients with autosomal recessive spinocerebellar ataxia. Clin Genet 97:532–535. https://doi.org/10.1111/cge.13669
Nakamura-Shindo K, Ono K, Koh K et al (2020) A novel mutation in the GBA2 gene in a Japanese patient with SPG46: a case report. eNeurologicalSci 19:100238. https://doi.org/10.1016/j.ensci.2020.100238
Kloth K, Cozma C, Bester M et al (2020) Dystonia as initial presentation of compound heterozygous GBA2 mutations: expanding the phenotypic spectrum of SPG46. Eur J Med Genet 63:103992. https://doi.org/10.1016/j.ejmg.2020.103992
Holla VV, Surisetti BK, Prasad S et al (2021) SPG46 due to truncating mutations in GBA2: two cases from India. Parkinsonism Relat Disord 82:13–15. https://doi.org/10.1016/j.parkreldis.2020.11.007
Lan M-Y, Lu C-S, Wu S-L et al (2022) Clinical and genetic characterization of a Taiwanese cohort with spastic paraparesis combined with cerebellar involvement. Front Neurol 13:1005670. https://doi.org/10.3389/fneur.2022.1005670
Cores Bartolomé C, Rubio Nazábal E, Sobrido MJ, Pérez Sousa C (2023) SPG46 spastic paraplegia due to GBA2 mutation: description of the first case in Spain. Neurologia (Engl Ed) 38:372–374. https://doi.org/10.1016/j.nrleng.2022.06.009
Scarpelli M, Zappini F, Filosto M et al (2012) Mitochondrial sensorineural hearing loss: a retrospective study and a description of cochlear implantation in a MELAS patient. Genet Res Int 2012:287432. https://doi.org/10.1155/2012/287432
Fancello V, Fancello G, Palma S et al (2023) The role of primary mitochondrial disorders in hearing impairment: an overview. Medicina (Kaunas) 59:608. https://doi.org/10.3390/medicina59030608
Sultana S, Stewart J, van der Spoel AC (2020) Truncated mutants of beta-glucosidase 2 (GBA2) are localized in the mitochondrial matrix and cause mitochondrial fragmentation. PLoS One 15:e0233856. https://doi.org/10.1371/journal.pone.0233856
Roshan Lal T, Sidransky E (2017) The spectrum of neurological manifestations associated with Gaucher disease. Diseases 5:10. https://doi.org/10.3390/diseases5010010
Bremova-Ertl T, Schiffmann R, Patterson MC et al (2017) Oculomotor and vestibular findings in Gaucher disease type 3 and their correlation with neurological findings. Front Neurol 8:711. https://doi.org/10.3389/fneur.2017.00711
Vitner EB, Futerman AH (2013) Neuronal forms of Gaucher disease. Handb Exp Pharmacol 405–419. https://doi.org/10.1007/978-3-7091-1511-4_20
Boer DEC, van Smeden J, Bouwstra JA, Aerts JMFG (2020) Glucocerebrosidase: functions in and beyond the lysosome. J Clin Med 9:736. https://doi.org/10.3390/jcm9030736
Körschen HG, Yildiz Y, Raju DN et al (2013) The non-lysosomal β-glucosidase GBA2 is a non-integral membrane-associated protein at the endoplasmic reticulum (ER) and Golgi. J Biol Chem 288:3381–3393. https://doi.org/10.1074/jbc.M112.414714
Sabbagh A, Pasmant E, Laurendeau I et al (2009) Unravelling the genetic basis of variable clinical expression in neurofibromatosis 1. Hum Mol Genet 18:2768–2778. https://doi.org/10.1093/hmg/ddp212
Jobling R, Ferrier RA, McLeod R et al (2011) Monozygotic twins with variable expression of Van der Woude syndrome. Am J Med Genet A 155A:2008–2010. https://doi.org/10.1002/ajmg.a.34022
Collins AL, Lunt PW, Garrett C, Dennis NR (1993) Holoprosencephaly: a family showing dominant inheritance and variable expression. J Med Genet 30:36–40. https://doi.org/10.1136/jmg.30.1.36
Kingdom R, Wright CF (2022) Incomplete penetrance and variable expressivity: from clinical studies to population cohorts. Front Genet 13:920390. https://doi.org/10.3389/fgene.2022.920390
Cavalli G, Heard E (2019) Advances in epigenetics link genetics to the environment and disease. Nature 571:489–499. https://doi.org/10.1038/s41586-019-1411-0
Skre H, Berg K (1977) Linkage studies on Marinesco-Sjøgren syndrome and hypergonadotropic hypogonadism. Clin Genet 11:57–66. https://doi.org/10.1111/j.1399-0004.1977.tb01279.x
Anttonen A-K, Mahjneh I, Hämäläinen RH et al (2005) The gene disrupted in Marinesco-Sjögren syndrome encodes SIL1, an HSPA5 cochaperone. Nat Genet 37:1309–1311. https://doi.org/10.1038/ng1677
Senderek J, Krieger M, Stendel C et al (2005) Mutations in SIL1 cause Marinesco-Sjögren syndrome, a cerebellar ataxia with cataract and myopathy. Nat Genet 37:1312–1314. https://doi.org/10.1038/ng1678
Krieger M, Roos A, Stendel C et al (2013) SIL1 mutations and clinical spectrum in patients with Marinesco-Sjogren syndrome. Brain 136:3634–3644. https://doi.org/10.1093/brain/awt283
Anttonen AK. Marinesco-Sjögren Syndrome. 2006 Nov 29 [Updated 2019 Jan 10]. In: Adam MP, Feldman J, Mirzaa GM, et al (eds) GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1192/
Aureli M, Gritti A, Bassi R et al (2012) Plasma membrane-associated glycohydrolases along differentiation of murine neural stem cells. Neurochem Res 37:1344–1354. https://doi.org/10.1007/s11064-012-0719-z
Boot RG, Verhoek M, Donker-Koopman W et al (2007) Identification of the non-lysosomal glucosylceramidase as beta-glucosidase 2. J Biol Chem 282:1305–1312. https://doi.org/10.1074/jbc.M610544200
Woeste MA, Stern S, Raju DN et al (2019) Species-specific differences in nonlysosomal glucosylceramidase GBA2 function underlie locomotor dysfunction arising from loss-of-function mutations. J Biol Chem 294:3853–3871. https://doi.org/10.1074/jbc.RA118.006311
Amory JK, Muller CH, Page ST et al (2007) Miglustat has no apparent effect on spermatogenesis in normal men. Hum Reprod 22:702–707. https://doi.org/10.1093/humrep/del414
van der Spoel AC, Mott R, Platt FM (2008) Differential sensitivity of mouse strains to an N-alkylated imino sugar: glycosphingolipid metabolism and acrosome formation. Pharmacogenomics 9:717–731. https://doi.org/10.2217/14622416.9.6.717
Marshall J, Sun Y, Bangari DS et al (2016) CNS-accessible inhibitor of glucosylceramide synthase for substrate reduction therapy of neuronopathic Gaucher disease. Mol Ther 24:1019–1029. https://doi.org/10.1038/mt.2016.53
Sardi SP, Viel C, Clarke J, et al (2017) Glucosylceramide synthase inhibition alleviates aberrations in synucleinopathy models. Proc Natl Acad Sci U S A 114:2699–2704.https://doi.org/10.1073/pnas.1616152114
Funding
Open access funding provided by Università degli Studi di Roma La Sapienza within the CRUI-CARE Agreement.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Declarations
This multicentric case series study was performed in accordance with the Declaration of Helsinki statements. Written informed consent and ethical approval (CE Lazio) were obtained. We thank the contribution of Ricerca Corrente 2023 of the Italian Ministry of Health. The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cioffi, E., Coppola, G., Musumeci, O. et al. Hereditary spastic paraparesis type 46 (SPG46): new GBA2 variants in a large Italian case series and review of the literature. Neurogenetics (2024). https://doi.org/10.1007/s10048-024-00749-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10048-024-00749-9