

Abstract
There are still essential aspects regarding cathodes requiring a comprehensive understanding. These include identifying the underlying phenomena that prevent reaching the theoretical capacity, explaining irreversible losses, and determining the cut-off potentials at which batteries should be cycled. We address these inquiries by investigating the cell's capacity and phase dynamics by looking into the transport properties of electrons. This approach underlines the crucial role of electrons in influencing battery performance, similar to their significance in other materials and devices such as transistors, thermoelectrics, or superconductors. We use lithium iron phosphate LFP as a case study to demonstrate that understanding the electrochemical cycling behavior of a battery cell, particularly a Li//LFP configuration, hinges on factors like the total local potentials used to calculate chemical potentials, electronic density of states (DOS), and charge carrier densities. Our findings reveal that the stable plateau potential difference is 3.42 V, with maximum charge and minimum discharge potentials at 4.12 V and 2.80 V, respectively. The study illustrates the dynamic formation of metastable phases at a plateau voltage exceeding 3.52 V. Moreover, we establish that determining the working chemical potentials of elements like Li and Al can be achieved by combining their workfunction and DOS analysis. Additionally, we shed light on the role of carbon black beyond conductivity enhancement. Through Density functional theory (DFT) calculations and experimental methods involving scanning Kelvin probe (SKP) and electrochemical analysis, we comprehensively examine various materials, including Li, C, Al, Cu, LFP, FePO4, Li0.25FePO4, polyvinylidene fluoride, and Li6PS5Cl. The insights derived from this study, which solely rely on electrical properties, have broad applicability to all cathodes and batteries. They provide valuable information for efficiently selecting optimal formulations and conditions for cycling batteries.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 4.0 license. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
The heavy reliance on fossil fuels and centralized electricity grids poses constraints on our daily lives while also giving rise to global tensions and ecological crises. In response to this situation, researchers worldwide are dedicating their efforts to combat pollution by harnessing and storing energy. By reducing material usage, opting for more sustainable and less scarce materials, and developing environmentally friendly solutions, ongoing research aims to ensure continuous progress [1].
Solar, wind, and other intermittent green energy sources require effective battery technologies for energy storage. Additionally, the transition towards sustainable mobility hinges on advancements in efficient energy storage. Among the most common cathode materials for lithium-ion batteries are the lithium transition metal oxides of the LiMO2 family (M = Co, Ni, Mn,...) and the lithium manganese spinel LiMn2O4 [2–4]. Decades of study have revealed some significant drawbacks of these cathodes when applied in lithium-ion devices: (1) the transition metal oxides are prone to structural instability during cycling, leading to safety issues, and (2) the lithium manganese spinel reacts with liquid electrolytes. In contrast, the lithium polyanion compound family (Lix My )(XO4)Z where M = Co, Mn, Fe, V, ..., X = S, Si, P, ... and, for example, x = 3, y = 2, z = 3, presents a more promising option. A standout candidate is lithium iron phosphate LiFePO4, or LFP, which was developed in 1997 by Padhi et al [5, 6] following the polyanion cathode classification introduced by Manthiram and Goodenough [7]. LFP has emerged as a potential cornerstone for the next generation of lithium-ion batteries [8]. Its significance stems from being an environmentally benign and low-toxicity cathode material, offering a blend of safety, reliable cycling, and a long cycle life [9]. Moreover, the raw materials are economically viable and readily accessible. LFP's main drawbacks are its relatively low ionic and electrical conductivities, which lead to sluggish kinetics and limit its broader applicability [10]. LFP is recognized for its olivine crystal structure [11].
It is noteworthy that LiFePO4 can form more than one metastable Li-poor phase during charging. The robust covalent bonding between oxygen and phosphorous ions contributes to structural stability even when fully charged in the FePO4 phase. A slight distortion in the framework during cycling yields a flat plateau at approximately 3.45 V versus time or Li concentration  in Lix
FePO4 [1]. Here, we demonstrate theoretically and experimentally that x = 0, FePO4, corresponds to a plateau at ∼3.42 V, and x = 0.25, to ∼3.54 V (Lix
FePO4) for a quasi-ideal cycling rate, and not accounting for the internal resistance. While the theoretical capacity of LFP material is 170 mAh g−1, achieving this value is a practical challenge. This difficulty arises because instead of reaching the fully delithiated or fully lithiated phase, an intermediate phase with x = 0.25 and less stable than FePO4 is often obtained, as illustrated by the equation
in Lix
FePO4 [1]. Here, we demonstrate theoretically and experimentally that x = 0, FePO4, corresponds to a plateau at ∼3.42 V, and x = 0.25, to ∼3.54 V (Lix
FePO4) for a quasi-ideal cycling rate, and not accounting for the internal resistance. While the theoretical capacity of LFP material is 170 mAh g−1, achieving this value is a practical challenge. This difficulty arises because instead of reaching the fully delithiated or fully lithiated phase, an intermediate phase with x = 0.25 and less stable than FePO4 is often obtained, as illustrated by the equation  compromising the effective capacity under experimental conditions.
compromising the effective capacity under experimental conditions.
The inherent stability of the FePO4 framework makes extracting oxygen atoms more challenging compared to other oxide cathode materials, ensuring excellent thermal stability up to 400 °C [12]. However, at lower temperatures, LFP materials exhibit insufficient capacity due to several factors: (1) the olivine structure restricts Li+ diffusion along the (010) direction, resulting in low ionic conductivity [13]; (2) the absence of a continuous FeO6 network leads to low electronic conductivity [14]; and (3) the phase-transforming lithiation-delithiation process during cycling impacts battery performance under severe/extreme conditions, resulting in the formation of unwanted phases, as mentioned previously [13].
The electronic properties of LFP have been widely debated in the literature, as the cause of its low electrical conductivity, commonly referred to as the 'band gap problem' [15], remains incompletely understood. The electronic band structure facilitates the analysis of electron transport in solid materials, a focal point of this study. As such, we present simulated electronic band structures and calculated electrical properties that align with existing literature and our experimental analysis.
Cui et al [16] reported a band gap of 3.78 eV for LFP without doping. supplementary information (SI) tables S1 and S2 outline various methods for calculating the LFP band gap. Furthermore, doping LFP facilitates electron transfer and enhances electrical conductivity. Nevertheless, we demonstrate that this gap is not the primary one. All electronic properties are corroborated through the plateau's potential and charge carrier density range during cycling and the cathode's capacity. Indeed, all electrochemical dynamics and intricacies can be elucidated through the electronic transport in LiFePO4, Li0.25FePO4, and FePO4.
This process begins with calculating chemical potential, corresponding to the Fermi level for electrically insulated phases (figures 1(a)–(c)). By determining the average potential leading to the corresponding phases' workfunction, we were able to locate the Fermi level with reference to electrons at rest at the surface in a vacuum. The theoretical simulations were performed admitting no surface potential arises by electrical contact with other species or by self-phenomena; in this case, the Fermi level or electrochemical potential corresponds to the chemical potential as shown in figure 1(c).
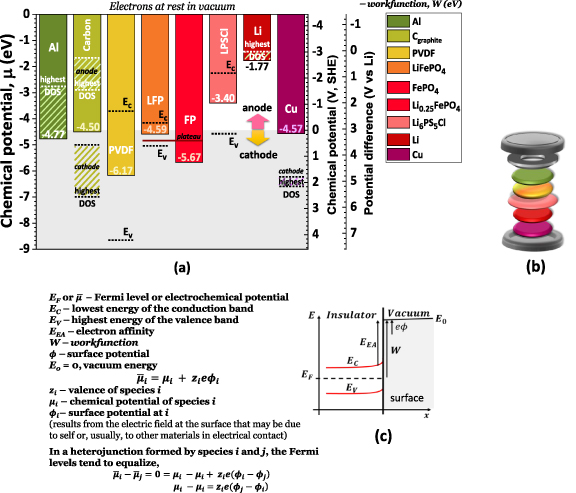
Figure 1. Summary of the (a) chemical potentials, workfunctions, electronic density-of-states (DOS), and band gap ( ) relevant to the materials used in this study; (b) schematic model of the materials studied and their hierarchical position in the cell; (c) representation of the energies and concepts that explain how the chemical potentials bias of a heterojunction is overcome by the accumulation of charge of opposite signs at the surfaces in electrical contact (conductor-insulator) or by exchange of electrons (conductor-conductor). Note: The highest DOS regions (diagonal strips) point to the working or 'effective' chemical potential and to the different roles a material may have in a battery cell, such as carbon that may be found in both the anode and cathode. The anode working chemical potentials of Li, Al, and carbon (graphite) match the highest DOS, as shown in [17]. All scales correspond directly: −4.44 eV/e = 0 V, SHE = 3.05 V vs Li0. All chemical potentials in the grey background color (<−4.44 eV) represent cathode-like materials, and in the white background color (>−4.44 eV) represent anode-like materials (with a small electron affinity,
) relevant to the materials used in this study; (b) schematic model of the materials studied and their hierarchical position in the cell; (c) representation of the energies and concepts that explain how the chemical potentials bias of a heterojunction is overcome by the accumulation of charge of opposite signs at the surfaces in electrical contact (conductor-insulator) or by exchange of electrons (conductor-conductor). Note: The highest DOS regions (diagonal strips) point to the working or 'effective' chemical potential and to the different roles a material may have in a battery cell, such as carbon that may be found in both the anode and cathode. The anode working chemical potentials of Li, Al, and carbon (graphite) match the highest DOS, as shown in [17]. All scales correspond directly: −4.44 eV/e = 0 V, SHE = 3.05 V vs Li0. All chemical potentials in the grey background color (<−4.44 eV) represent cathode-like materials, and in the white background color (>−4.44 eV) represent anode-like materials (with a small electron affinity,  ).
).
Download figure:
Standard image High-resolution imageA survey of existing literature emphasizes the extensive scientific efforts dedicated to interfacial engineering in systems employing LFP as an active cathode material [18–20]. Generally, the LFP slurry is formulated by blending the active material with carbon-conductive powder and a polymer binder in an 8:1:1 weight ratio [21]. Each component serves a specific purpose: (1) carbon material enhances electrical conductivity; (2) the polymer binder ensures material cohesion, polarizes the cathode, and facilitates ionic conduction due to its high dielectric constant; and (3) the active material itself provides theoretical capacity.
This work investigates the electrochemical roles of various components, including LFP, polyvinylidene fluoride (PVDF), Cblack, Li6PS5Cl (LPSCl) argyrodite—LPSCl solid electrolyte, Li, and current collectors of Al and Cu. While this study centers on Li//LFP batteries, its applicability extends to Li+-ion and solid-state batteries. It focuses on the cathode; a similar analysis may be developed for any other active material.
We investigate the intrinsic surface and physicochemical properties of LFP in an all-solid-state cell composed of Al/LFP/Cu. This approach enables us to obtain experimental surface chemical potentials and chemical potentials and compare them with simulated correspondents. By incorporating other cell components into this study, we identify previously unknown roles that significantly influence the optimized behavior of the cathode and, therefore, of the battery cell.
A traditional cathode slurry was prepared and evaluated using experimental techniques such as cyclic voltammetry (CV), electrochemical cycling, and SKP, shedding light on the chemical potential and conductor behavior of carbon, PVDF, and the current collectors Cu and Al. A similar cell configuration was employed to explore the feasibility of a cathode bilayer using the solid electrolyte LPSCl, which allowed us to analyze the chemical potential of each material and its ability to balance surface chemical potentials in mixtures (e.g. cathode) and heterojunctions.
The role of carbon was also closely examined, revealing its critical and versatile behavior in both cathodes and anodes. The transport of electrons and equalization of surface chemical potentials heavily rely on a minimal amount of carbon black in the mixture as low as 3 wt%, as demonstrated hereafter. Importantly, this role is analyzed here for the first time while correlated with its chemical potential and maximum density of states (DOS). The carbon's versatility and possibility of application in both anode and cathode are shown to be directly related to similar values of the maximum DOS for anode and cathode-like chemical potentials corresponding to densities of states accessible to electrons and holes, respectively, as shown in figure 1(a).
Hereafter, we will show the role of each species composing a solid-state battery for the first time, solely relying on their relative chemical potential, DOS, and charge carrier density.
In summary, we show that if the phases that may form during the cycling of the active material are known, by simulating the chemical potential vs charge carriers density, we can determine (1) the open circuit voltage (OCV); (2) the stable plateau voltage; (3) the metastable plateau voltage (if there is one); (4) the best minimum discharge voltage; (5) the best maximum charge voltage; (6) the fast and the equilibrium cycling curves and their (6) corresponding theoretical capacities.
By calculating the capacitance ( , using the charge carriers density and the chemical potentials vs Li0/Li+, the two-phase reaction stable plateau is determined and highlighted that the electrical double layer capacitors (EDLCs) do not vary at the plateau voltage as the chemical potentials of all the stable phases in the anode and cathode are constant. Using the DOS, it is also possible to determine the effective chemical potentials of the electrodes and collectors materials such as Li, C, Cu, and Al and their possible roles as anodes and/or cathodes. All of the following are based on the simulation of the total local potential and electrical transport properties, not accounting for the impedances as they vary from cell to cell.
, using the charge carriers density and the chemical potentials vs Li0/Li+, the two-phase reaction stable plateau is determined and highlighted that the electrical double layer capacitors (EDLCs) do not vary at the plateau voltage as the chemical potentials of all the stable phases in the anode and cathode are constant. Using the DOS, it is also possible to determine the effective chemical potentials of the electrodes and collectors materials such as Li, C, Cu, and Al and their possible roles as anodes and/or cathodes. All of the following are based on the simulation of the total local potential and electrical transport properties, not accounting for the impedances as they vary from cell to cell.
Experimentally, recurring to SKP, we obtain the role and importance of each material's contribution to the electronic and ionic conductivities in the cell, as well as its contribution to the OCV and cell's electrochemical performance.
2. Results and discussion
An initial characterization of the materials (SI, Methods) was established by calculating the workfunction of the simulated surfaces and the electronic band structure, DOS, and charge carriers of the bulk structure of all the materials directly involved in the performance of a Li//LFP solid-state battery (figures 1(a) and (b)). The conclusions herein extend to the Li-ion LFP battery with a graphite anode.
The workfunction is the minimum thermodynamic work needed to remove an electron from the Fermi level to a point immediately outside the solid surface (figure 1(c)), admitting the electrons at rest when reaching the surface and corresponding to the electrochemical potential and to the chemical potential with no surface potential ( , due to self-charge surface phenomena or the electrical presence of other materials [22]. The workfunctions of aluminum, carbon, copper, and lithium were calculated, and the values obtained were 4.77 eV, 4.57 eV, 4.50 eV, and 1.77 eV, respectively (figures 1(a) and 2(a)). A low workfunction is generally associated with a material that is able to provide an efficient electron ejection. Copper, compared to aluminum, is shown here to have a smaller workfunction; however, in practical applications, Al may perform as an anode, and while lithium is applied as an anode material, copper is usually used as a negative current collector and may be used as cathode. The latter shows that the effective chemical potentials also depend on DOS, and Cu has fewer DOS for electrons than Al, as required for the anode functionality.
, due to self-charge surface phenomena or the electrical presence of other materials [22]. The workfunctions of aluminum, carbon, copper, and lithium were calculated, and the values obtained were 4.77 eV, 4.57 eV, 4.50 eV, and 1.77 eV, respectively (figures 1(a) and 2(a)). A low workfunction is generally associated with a material that is able to provide an efficient electron ejection. Copper, compared to aluminum, is shown here to have a smaller workfunction; however, in practical applications, Al may perform as an anode, and while lithium is applied as an anode material, copper is usually used as a negative current collector and may be used as cathode. The latter shows that the effective chemical potentials also depend on DOS, and Cu has fewer DOS for electrons than Al, as required for the anode functionality.
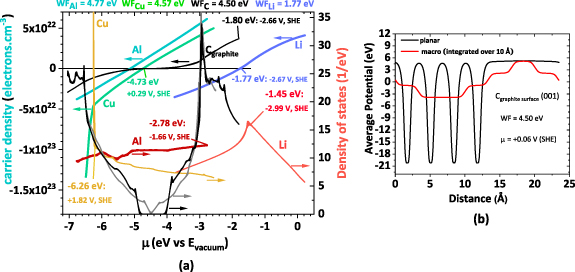
Figure 2. DFT simulations (a) for carrier density (left scale) and DOS (right scale) vs. chemical potential (μ) for all the employed conductors (from 50 to 300 K) and (b) carbon (graphite surface) workfunction and chemical potential for a (001) surface considering no surface potential ( . Calculated from the average potential integrated over 10 Å. Note: Carbon was simulated in the graphite crystal structure, starting from two slightly different variations of the structure, both optimized before simulating the electrical properties. The arrows point to the correspondent scale. Scales: −4.44 eV/e = 0 V, SHE = 3.05 V vs Li0.
. Calculated from the average potential integrated over 10 Å. Note: Carbon was simulated in the graphite crystal structure, starting from two slightly different variations of the structure, both optimized before simulating the electrical properties. The arrows point to the correspondent scale. Scales: −4.44 eV/e = 0 V, SHE = 3.05 V vs Li0.
Download figure:
Standard image High-resolution imageIn solid-state Physics, the DOS is a measure of the number of electronic states at a given chemical potential available for electrons to occupy [23] (figure 2(a)). Furthermore, DOS can be used to predict the material's ability to transport electrons or to create holes as available states are necessary. A high value of DOS at the conduction band is found in materials more available to transport electrons; accessible electronic states at high energy levels allow electrons to move freely with less resistance. A high value of DOS at the valence band generally indicates the presence of many available electronic states at low energy levels. Lithium, aluminum, and copper are metals; in metals, the valence band is not filled and coincides with the conduction band, or the valence and conduction bands overlap, allowing electrons to move freely in the material. In all these metallic materials, the value of DOS in the conduction band is generally high, resulting in good electrical conductivity.
Density functional theory (DFT) simulations were performed to predict the behavior of the employed materials, considering the relationship between the chemical potentials (µ), carriers density, and DOS (figure 2).
At first sight, by charge carrier and DOS analysis, it is possible to anticipate that carbon allows the electrons to drift in a larger specter than the other conductors. Additionally, it enables the existence of both electron and hole transport. In other words, it is more likely that carbon-based materials allow the equalization of surface chemical potentials when in contact with other species in a heterojunction cell owing to their chemical potential (figure 2(b)) and very high DOS corresponding to both charge carriers (electrons and holes). This intrinsic characteristic explains why carbon is empirically widely used with active materials to produce both positive and negative electrodes.
Copper and lithium tend to have higher DOS when compared with aluminum, which shows very similar DOS for electrons and holes, explaining its high capacity to deliver a broad range of surface chemical potentials, as pointed out by the authors previously [24, 25].
Notably, the conjunction of the highest DOS with a high charge carrier density defines the working chemical potential, which may be severely shifted from the calculated workfunction at null surface potential. The latter conclusion is demonstrated for Al with a maximum DOS value of −1.66 V (SHE) as opposed to the estimated potential μ (Al) = +0.33 V (SHE) and Li with a maximum DOS value of −2.99 V (SHE) as opposed to the calculated potential μ (Li) = −2.67 V (SHE), both for electrons (figure 2(a)) and indicating anode-like behavior. The experimental working absolute potentials were published in 1986 by IUPAC and presented by Trasatti [17], μ (Al) = −1.66 ± 0.02 V (SHE) and μ (Li) = −3.05 ± 0.02 V (SHE).
The discrepancy between the experimental workfunctions and 'working' chemical potentials was previously observed for Al and Li and attributed to self-surface potential phenomena [22, 26]. However, here it is shown for the first time that the 'working' chemical potential has an origin in the overlap between the workfunction and a high DOS that results in a shift of electrons towards higher energies, corresponding to the material working state, for example, as a negative electrode like in the case of Aluminum (figure 2(a)).
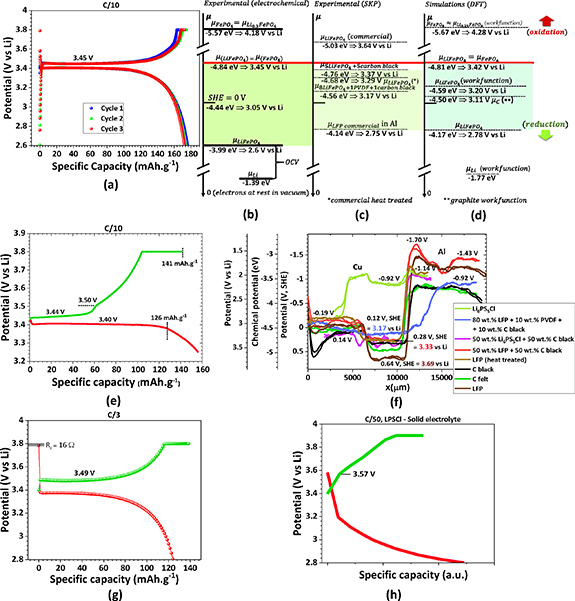
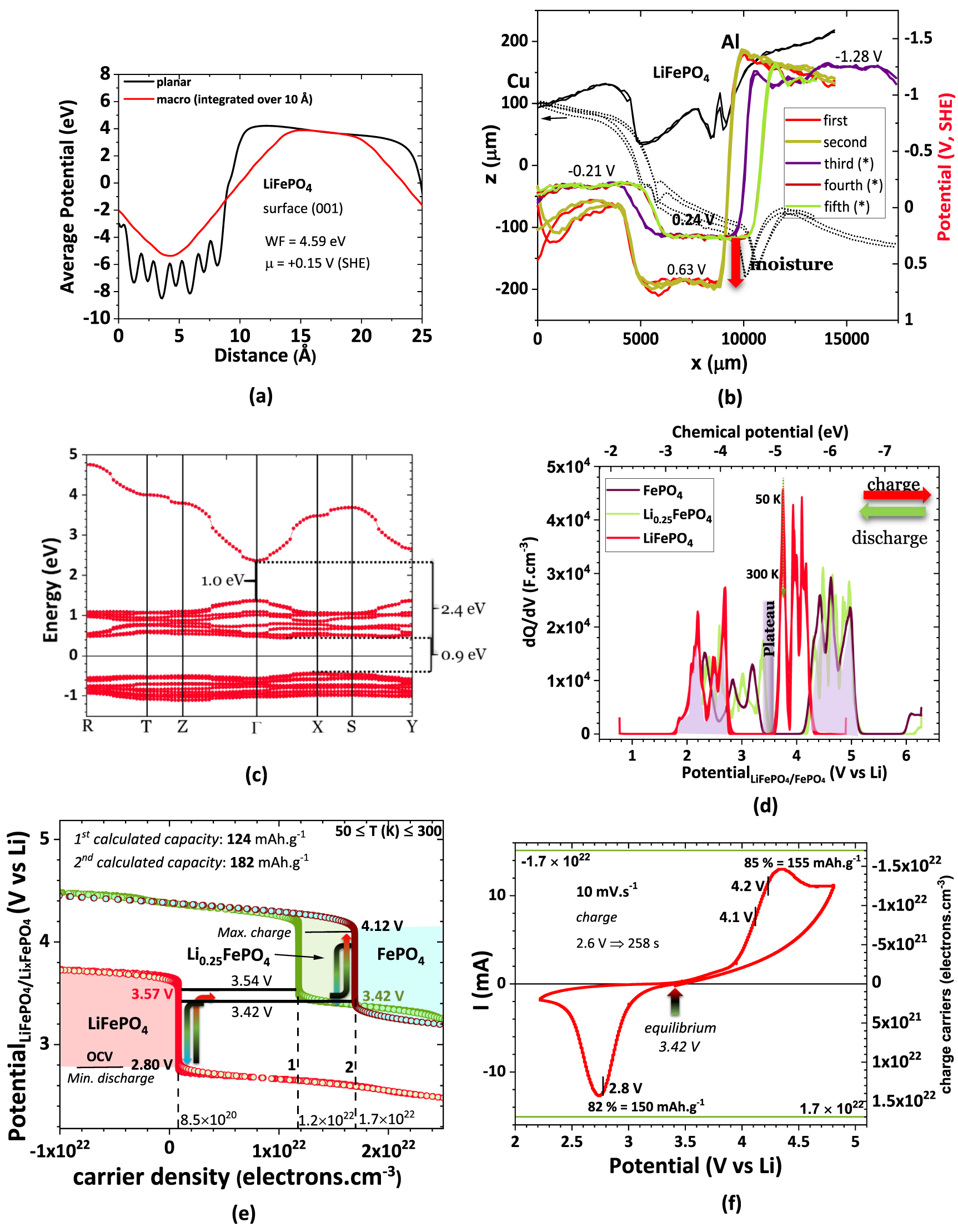
The FePO4 and Li0.25FePO4 have the same chemical potential (SI, figure S2(b)), corresponding to a higher workfunction than the LiFePO4, as expected because these phases perform as cathodes and are stable when the cell is charged (figure 1). Recurring to ab initio simulations, it was possible to obtain the theoretical workfunction of LFP of 4.59 eV in the (001) direction, corresponding to a chemical potential of +0.15 V (SHE) (figure 3(a)), not accounting for any surface potential ( V).
V).

Figure 3. DFT calculations of (a) LFP workfunction and surface chemical potential corroborating the (b) SKP profile of the Cu/LFP/Al cell, showing the difference between the moisty and dry LFP. Simulation of the (c) electronic band structure vs. the directions in the Brillouin zone for LFP with signalized band gaps; (d) differential capacitance analysis of three FePO4, Li0.25PO4 and LiFePO4 phases vs. potential, highlighting the plateau where no differential capacitance variation for two phases (Li-rich and Li-poor) corresponds to the phase equilibria where the chemical potentials of the phases remain constant; (e) phase diagram or cycling profile of FePO4, Li0.25PO4 and LiFePO4 phases, obtained from chemical potentials vs. charge carrier density highlighting and backing up the maximum charge and minimum discharge and equilibrium potentials, as well as the capacity of the cell and the Li0.25FePO4 metastable phase; (f) experimental cyclic voltammetry for a Li//LFP cell with 1M LiPF6 EC/DMC electrolyte showing the number of charge carriers here corresponding to electrons (+ carriers) and Li+/holes (- carriers) and the potential pinpoints in (e); voltage rate 10 mV s−1 and RT = 20 °C. Note: to calculate the charge carrier density from the experimental data we have used LFP simulated density of 3.924 g cm−3. Scales: −4.44 eV/e = 0 V, SHE = 3.05 V vs Li0.
Download figure:
Standard image High-resolution imageThe PVDF has the lowest chemical potential value of −6.17 eV or +1.7 V (SHE), not accounting for any surface potential ( V) (SI, figure S2(a)). This polymer is piezoelectric, meaning that this material can polarize when poled, for example, by mechanical stimuli [27, 28]. In this work, the PVDF was difficult to polarize without being poled or prepared in the traditional cathode slurry.
V) (SI, figure S2(a)). This polymer is piezoelectric, meaning that this material can polarize when poled, for example, by mechanical stimuli [27, 28]. In this work, the PVDF was difficult to polarize without being poled or prepared in the traditional cathode slurry.
The LPSCl electrolyte shows an electronic band gap with ∼2.3 eV obtained by ab initio simulations herein and 2.14 eV in [29], thus classifying the electrolyte as a semiconductor. Alternatively, the not-so-large workfunction is translated into an anode-like potential (figure 1(a)) with a not-as-low electrical conductivity as other electrolytes with more intrinsic insulator characteristics. This electrolyte has been used in different studies [30–32] with recognized advantages [33–36].
It is possible that an interphase forms at the interface when the electrolyte is in the presence of Li. With the natural tendency to equalize the electrochemical potentials between the anode and the electrolyte, electrons may tunnel to the conduction band of the LPSCl electrolyte at a lower energy level, reducing it until forming an insulator interphase with a conduction band with higher energy than the chemical potential of Lithium (figure 1). When the latter happens, the tunneling to the electrolyte surface ceases.
A question arises: how does the formation of interphases, spurious reactions, or moisture absorption affect the cell's voltage?
Due to the semiconductor properties of LFP, its capabilities to equalize the surface chemical potentials with other species in contact are not expected to be as efficient as with a conductor cathode (figures 3(a), (c), SI, S4, and S5). The latter is observed in a heterojunction cell prepared by placing LFP powder in the gap between copper and aluminum metals (Cu/LFP/Al). The circuit was closed by connecting the copper to the SKP equipment. As anticipated, a sharp quantum well is unequivocal, which leads to the conclusion that the olivine structure does not allow the tunneling of electrons from the metal electrodes to provide surface potential compensation [37].
While the first and second experiments were performed with LFP that was not kept in an air-dry environment, for subsequent measurements, the LFP powder was dried in a vacuum oven for two hours at 70 °C to guarantee that no moisture would subsist in the as-stored condition [38]. The SKP measurements were carried out in an air-dry environment with below zero dew point.
In figures 3(b), SI, and S7, the moisture effect on the electrochemical behavior is shown to be giant; to our knowledge, this seems to be the first quantitative practical experimental result corroborating existing simulations and theories [39–41]. The quantum well increases by almost 0.4 V, declining even more the capacity of complete equalization with either electrode surface chemical potentials (figure 3(b)). No polarization leading to differently charged regions was found in the LFP.
What are the pinpoints in the Li//LFP battery-cell electrochemical cycling (maximum, minimum, plateau potential, and capacity), and how are they related to electronic band structure and charge carriers' transport?
Literature suggests [42–44] mainly two different approaches when defining the formation of the phases throughout the lithiation and delithiation processes, as already mentioned herein: a simpler one that considers the full lithiation and delithiation process and another which comprises different percentages of lithium intercalation/deintercalation. The cycling process is one of the highest conditioning factors when handling an LFP-based battery [45, 46]. It is mandatory to understand the limits for these two processes, which are linked with LiFePO4 and FePO4 phases [47] and, depending on the rates, Li0.25FePO4. Aiming to understand this problem, DFT simulations were performed to associate the average total local potentials, from which the chemical potentials were calculated, the charge carriers' density (figures 3(c)–(f)), and the differential capacitance due to the accumulation of charged species.
The electron transport that reflects the electronic band structure in figure 3(c) is expressed in the differential capacitance vs. chemical potential (Li0/Li+) in figure 3(d).
When two phases are in equilibrium in a two-phase region, the chemical potentials of both phases are equal, and none of the phases varies in the number of charge carriers. Therefore, the electric double-layers (EDLC) formed spontaneously to equalize the electrochemical potentials, or Fermi levels, remain with constant capacitance at equilibrium. What varies is the amount of each phase, obeying the lever rule. The range of potentials where an equilibrium between the two most stable phases, LiFePO4 and FePO4, may happen is small and corresponds to 3.42–3.57 V.
When the discharged cell is charged in quasi-equilibrium conditions, FePO4 forms at 3.42 V, corresponding to the plateau (1−x) LiFePO4 + x FePO4, where  . The equilibrium evolves when LiFePO4 runs out for
. The equilibrium evolves when LiFePO4 runs out for  . If, however, the equilibrium conditions are not achieved because, for example, the C-rate is too high, then the less stable Li0.25FePO4 phase may form. The equilibrium will not happen or will be established at higher energy, 3.54–3.57 V vs. Li0 (figures 3(d), (e) and SI, S3). Above 3.57 V, no equilibrium plateau should be observed, and LiFePO4 transforms into Li0.25FePO4 directly (figures 3(d), (e) and SI, S3), failing even to achieve 124 mAh g−1 figure 3(e) (1). The 182 mAh g−1 figure 3(e) (2) capacity is reached when all LFP is transformed into FePO4. The difference between the 170 mAh g−1, the maximum theoretical capacity, and the latter is due to the simulation parameters for LFP and FePO4, including the simulated crystal and electronic structures, surfaces, workfunctions, chemical potentials, electronic transport properties, and density. The maximum deviation is therefore calculated to be 7%.
. If, however, the equilibrium conditions are not achieved because, for example, the C-rate is too high, then the less stable Li0.25FePO4 phase may form. The equilibrium will not happen or will be established at higher energy, 3.54–3.57 V vs. Li0 (figures 3(d), (e) and SI, S3). Above 3.57 V, no equilibrium plateau should be observed, and LiFePO4 transforms into Li0.25FePO4 directly (figures 3(d), (e) and SI, S3), failing even to achieve 124 mAh g−1 figure 3(e) (1). The 182 mAh g−1 figure 3(e) (2) capacity is reached when all LFP is transformed into FePO4. The difference between the 170 mAh g−1, the maximum theoretical capacity, and the latter is due to the simulation parameters for LFP and FePO4, including the simulated crystal and electronic structures, surfaces, workfunctions, chemical potentials, electronic transport properties, and density. The maximum deviation is therefore calculated to be 7%.
It is noteworthy to highlight how the differential capacitance,  varies out of the equilibrium, denoting the ability of the active cathode material to store charge and be reduced or oxidized (figure 3(d)).
varies out of the equilibrium, denoting the ability of the active cathode material to store charge and be reduced or oxidized (figure 3(d)).
From these results, some conclusions should be extracted: (1) the reaction  is the most stable, and the constant number of charge carriers of FePO4 and LiFePO4 suggests the discharge and charge limits should be 2.80 and 4.12 V, corresponding to total capacity; (2) the lowest the equilibrium potential in the range of 3.42–3.57 V, the highest the capacity of the battery-cell; (3) a flat plateau occurs where two curves (from Li-rich and Li-poor phases) overlap at
is the most stable, and the constant number of charge carriers of FePO4 and LiFePO4 suggests the discharge and charge limits should be 2.80 and 4.12 V, corresponding to total capacity; (2) the lowest the equilibrium potential in the range of 3.42–3.57 V, the highest the capacity of the battery-cell; (3) a flat plateau occurs where two curves (from Li-rich and Li-poor phases) overlap at  , corresponding to the two-phase equilibrium in the phase diagram.
, corresponding to the two-phase equilibrium in the phase diagram.
In ideal CV, the constant current lines (horizontal) in the stability range of the LiFePO4 and FePO4 phases should correspond to a difference in charge carriers equivalent to the full cell's capacity as shown in figure 3(e). Below 2.80 V and above 4.12 V, an almost rectangle angle should be observed (figure 3(e)), corresponding to the reduction and oxidation of the LiFePO4 and the FePO4 phases.
However, in figure 3(f), the cell's capacity was obtained out of equilibrium, but still, where the differential capacitance is  as shown in figure 3(d). Therefore, the capacity at the maximum charge carriers (holes and electrons) corresponded to 155 and 150 mAh g−1, consistent with the charge and discharge, respectively. Finally, the capacity is calculated from the number of charge carriers obtained from the current and charging time; the potential rate allows for estimating the charging or discharging time. We point out that at constant current (constant number of charge carriers), the differential resistance
as shown in figure 3(d). Therefore, the capacity at the maximum charge carriers (holes and electrons) corresponded to 155 and 150 mAh g−1, consistent with the charge and discharge, respectively. Finally, the capacity is calculated from the number of charge carriers obtained from the current and charging time; the potential rate allows for estimating the charging or discharging time. We point out that at constant current (constant number of charge carriers), the differential resistance  tends to be infinite, corresponding to the stability of the phases.
tends to be infinite, corresponding to the stability of the phases.
Suppose pure LFP is employed in a cathode. In that case, a little intercalation process is expected owing to its relatively low ionic and electric conductivities in its stability range of potentials (SI, figure S4). After testing the LFP powder itself, it was assessed the effect of carbon-black in the powder mixture (figures 4(a) and (b)). Then, a cathode slurry was also prepared with the traditional 8:1:1 proportion, as shown in figure 4(c).

Figure 4. SKP scans (a) 2D potential; (b) topography (CTM) and SKP measurement profiles corresponding to a Cu/50 wt.% LFP + 50 wt.% Cblack/Al cell showing the equalization of the Cu with the mixture and negatively charged plasmons solitons at the mixture's surface; (c) SKP profiles of the Cu/10 wt.% CBlack + 10 wt.% PVDF + 80 wt.% LFP/Al and Cu/3 wt.% CBlack + 40 wt.% LPSCl + 57 wt.% LFP/Al cells showing little difference between the surface chemical potentials in both cells, the only representative difference being the accumulation of negative charge at the interface mixture with LPSCl/Al, a characteristic of the solid electrolyte. Topography (CTM) and SKP profiles of (d) LFP commercial cathode showing the Al foil equalizing its surface chemical potential through the mixture deposited on Al. DFT calculation of (e) Li6PS5Cl workfunction and chemical potential for the (100) surface. Topography (CTM) and SKP scans of (f) Li6PS5Cl electrolyte in a Cu/Li6PS5Cl/Al cell and (g) in a mixture with carbon in Cu/50 wt.% Li6PS5Cl + 50 wt.% CBlack/Al cell. When CBlack is present, the transport of electrons and holes and the equalization of the chemical potentials Cu/50 wt.% Li6PS5Cl + 50 wt.% are observed. CBlack assumes the chemical potential of Cu when electrically insulated. If just the LPSCl is present, the electrolyte can align both Cu and Al at the same potential, bringing Cu's surface potential up more than 1 V at the interface, extending 2 mm to the Cu bulk. A sharp, negatively charged barrier is observed on both ends at the interfaces. Scales: −4.44 eV/e = 0 V, SHE = 3.05 V vs Li0.
Download figure:
Standard image High-resolution imageConversely to 100% Cblack (SI, figure S6), as described hereafter, 50 wt.% LFP + 50 wt.% Cblack aligns its surface chemical potential through the Cu by electron transport in the Cu/50 wt.% LFP + 50 wt.% Cblack/Al cell. In figures 4(a) and (b), the negatively charged plasmons (solitons) are observed and part of their dynamics [24, 25]. The cell transports electrons through the Cu/50 wt.% LFP + 50 wt.% Cblack surface (figures 4(a)and (b), SI, figures S8(a) and (b)).
To consolidate the most relevant conclusions so far, a new cell was assembled with 80 wt.% LFP, 10 wt.% CBlack and 10 wt.% PVDF and Cu and Al electrodes. Initially, in a slurry form, the traditional cathode dried out in the cell's gap before the experiment (SI, methods). The results are shown in figures 4(c), SI, S9(a) and (c).
PVDF polymer was used as a binder since it can bind all electrode components through interactions, from Van der Waals to electrostatic forces [48]. PVDF polymer has a high dielectric constant and a workfunction of 6.17 eV (SI, figure S2(a)). Some SKP tests were carried out, but the signal saturated very quickly, at ∼+5 V, which explains why PVDF topography and SKP plots were not included. Therefore, this possibly indicates that the PVDF does not polarize just by the action of the electric field created by the difference in Fermi levels between Al and Cu.
The Cu/80 wt.% LFP + 10 wt.% CBlack + 10 wt.%PVDF/Al and Cu/57 wt.% LFP + 40 wt.% LPSCl + 3 wt.% CBlack/Al cells show very similar behavior at the heterojunctions and interfaces and a similar bulk surface potential to the Cu/50 wt.% LFP + 50 wt.% Cblack/Al cell (figures 4(b) and (c)).
Three percent of CBlack is enough to equalize the surface chemical potential of a mixture containing 3 wt.% CBlack (+0.06, SHE) + 47 wt.% LPSCl (−1.04 V, SHE) + 50 wt.% LFP (+0.15 V, SHE), with the Cu at +0.10 V, SHE. The alignment of the chemical potential of the mixture with the Cu electrode is as efficient as the one obtained with a mixture with 10 wt.% CBlack (+0.06 V, SHE), 10 wt.% PVDF (+1.73 V, SHE) + 80 wt.% LFP (+0.15, SHE) at +0.14 V, SHE as shown in figure 4(c).
In SKP experiments with just 50 wt.% LPSCl + 50 wt.% LFP, the mixture does not equalize its surface chemical potential and shows zones of positive and negative saturation (SI, figure S10).
In the commercial cathode with 10 wt.% CBlack + 10 wt.% PVDF + 80 wt.% LFP deposited on Al, the aluminum brings the chemical potential of the cathode down from 3.17 to 2.77 V vs. Li0 (figures 4(c) and (d), SI, figures S9(b), and (d)). The latter potential is similar to the OCV of a Li//LFP battery cell.
A concluding study was carried out to study the applicability of bilayer cathode-solid electrolyte composite. SKP experiments were performed after assembling cells with Cu/LPSCl/Al, Cu/50 wt.% LPSCl + 50 wt.% LFP/Al, and Cu/50 wt.% LPSCl + 50 wt.% CBlack/Al. Using DFT simulations for LPSCl, the material's workfunction and, subsequently, the chemical potential was calculated, obtaining  and
and  , respectively, as shown in figure 4(f). Additionally, the electronic band gap of the electrolyte was also estimated, as pointed before, as 2.30 eV, indicating that the material behaves as a semiconductor.
, respectively, as shown in figure 4(f). Additionally, the electronic band gap of the electrolyte was also estimated, as pointed before, as 2.30 eV, indicating that the material behaves as a semiconductor.
To test the theoretical results experimentally, an SKP analysis was performed on a Cu/LPSCl/Al cell, shown in figure 4(f). As with other electrolytes studied by the authors [25], LPSCl allows the heterojunctions Cu/LPSCl and LPSCl/Al to equalize their surface chemical potentials at the same potential. However, in figure 4(f), it is shown that not only the latter phenomenon happens, but the Cu's chemical potential varies from −0.16 to −1.23 V in a step >2 mm away from the interface with the electrolyte. This singularity is observed by eyesight as the copper changes color in this >2 mm thick region. After the experiment, the electrolyte was removed, and the Cu showed the same surface chemical potential as before.
As pointed out, the LPSCl electrolyte is a semiconductor that shows solitons at the interface with the metals, corresponding to negatively charged species (figures 4(c) and (f)). The two accentuated standing peaks (solitons plasmons) may correspond to the electrons tunneling between the surface of the electrodes and the electrolyte, and this effect may be essential for the success of this electrolyte in all-solid-state batteries. However, when CBlack allows the conduction, plasmons may be identified in the bulk mixture surface 50 wt.% LPSCl + 50 wt.% CBlack, presenting alternate negatively and positively charged solitons. Another feature of the mixture is that the surface chemical potential shows a wavy-like polarized character after the first and second cycles. Essentially, the CBlack -LPSCl network creates electrical conductor paths, helping electrons to flow from one interface to the other, thus aligning the surface chemical potential of the Cu with the LPSCl and losing the alignment of this heterojunction with the Al (figure 4(f) vs figure 4(g)).
The final question: How should LFP-based cathodes and corresponding cells be optimized, tested, and conditioned?
Figures 5(a)–(f) show the extreme coherence between electrochemical cycling, SKP, and theoretical DFT simulations. At higher absolute potentials referenced to the electrons at rest in a vacuum, the OCV of a Li//LFP cell agrees with the surface chemical potential of the commercial cathode and with the simulated potential at which the LiFePO4 ceases to be a stable phase as it oxidizes (2.78–2.80 V). The simulated workfunction of the LFP, calculated from the total local potential of the LiFePO4 and specifically from the average potential integrated over 10 Å (figure 3(a)), is similar to the surface chemical potential of the plain LFP commercial powder and its mixture with CBlack, and with PVDF and CBlack, prepared in the traditional configuration with no Al current collector (figures 5(a) and (f)).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 5. Pinpoints in the (a) and (b) electrochemical cycles of a Li//FePO4 battery-cell in comparison with (c) SKP data and (d) DFT simulations of the chemical potentials; (e) electrochemical cycle at C/10 for a Li//LFP liquid electrolyte battery cell highlighting the departure from equilibrium at 3.50 V leading to the formation of Li0.25FePO4 as shown in figure 3; (f) SKP results for the surface chemical potentials showing the roles of the materials in the mixtures; the SKP data shows that in the surface away from the interfaces, the surface chemical potentials match the chemical potentials of the electrically insulated materials; (g) electrochemical cycle at C/3 (0.93 mA) for a Li//LFP liquid electrolyte battery cell, where the distance between the charge and discharge plateau is mainly related with the metastable phase formed (Li0.25FePO4), which is responsible for the plateau deviating immediately from 3.40 to 3.49 V and for the 125 mAh g−1 discharge capacity (cut off 2.8 V); the internal resistance is Ri = 16 Ω; (h) electrochemical cycle of a cell with solid-electrolyte Li/LPSCl/LFP at C/50 (0.008 mA) not showing plateau voltage; the cell charge evolves immediately to a potential above the equilibrium plateau range (3.57 V); the cell does not behave as a battery; it behaves as a capacitor with the cycling phenomena happening at the surface and the capacity reducing to less than 1 mAh g−1. The theoretical capacity and I ⩾ 1 μA were the constant voltage charging limits imposed.
Download figure:
Standard image High-resolution image{kind=link}
In the cycle in figure 5(e), corresponding to a Li//LFP battery cell with liquid electrolyte (1M LiPF6 EC:DMC), the cell starts charging at the equilibrium potential of 3.44 V, as the literature suggests [12, 49]. Subsequently, the system evolves to a 3.50 V charging voltage, demonstrating the formation of Li0.25FePO4 in agreement with a capacity reduction, as shown in figure 3(e).
The LPSCl solid electrolyte in figure 5(f) shows the ability to raise the chemical potential of Cu to equalize it with Al at −1.14 V (SHE), as discussed previously. The electrolyte away from the interfaces, in the quantum well, remains at −0.92 V, which is in agreement with the −1.04 V calculated from the simulated properties, using DFT, and with the SKP scans in figure 4(f) where the chemical potential reaches −1.02 V. When LPSCl is mixed with CBlack, the electrolyte deviates completely from its profile. In this case, it is possible to align the chemical surface potentials owing to the carbon capacity to transport electrons at the surface and bulk. The electronic solitons are no longer seen at the interfaces with this mixture.
The carbon chemical potential was calculated as +0.06 V (SHE) in the graphite allotrope. However, the carbon felt shows a surface chemical potential of +0.44 V, and the plain carbon black +0.33 V, which is maybe due to not being fully crystalline and also to the electrostatic effect observed in the powders; the powders of CBlack repeal each other when introduced in the ∼4 mm-thick confinement space between the Cu and Al. When CBlack is mixed with the LFP or even with LPSCl, the chemical potential shown is higher than the CBlack by itself, +0.12 and +0.13 V (SHE), respectively, and the plasmonic conduction is observed at least in the first measurements. All these chemical potentials were shown to be possible in figure 1(a) when CBlack is not charged with Li, working as a cathode-like material.
The role of CBlack is paramount, not only as a conductor to enable electron transport but to align the surface chemical potential of the semiconductors and insulator, LFP, LPSCl, and PVDF tested herein. Suppose only LFP, PVDF, or LFP and LPSCl compose the mixture (SI, figure S10). In that case, the mixture cannot equalize its surface chemical potentials with the electrodes, and polarized regions saturated at ±5 V (SHE) are observed.
The versatile role of carbon is likely due to its DOS, which is high for electrons and holes compared to other conductors and with no or almost no bandgap and lack of discontinuities in the DOS profile. The LFP semiconductor, with a small 0.9 eV band gap and high DOS for both electrons and holes, shows a pronounced discontinuity on its DOS profile and cannot equalize the surface chemical potentials by itself.
The results in figure 5(f) reveal their importance; the traditional cathode mixture with LFP, CBlack, and PVDF shows a soft surface chemical potential transition in all the scanned areas, denoting the interface's resistance reduction. This reduction is more pronounced on the Cu side but is still forgiven on the Al heterojunction side. The capacity to equalize the chemical potential is due to the electron drifting caused by the carbon conductive element and the binding effect provided by PVDF, allowing the combination of the materials and thus providing enhanced electrical properties. The PVDF effect was found to be binder-like, not interfering in equalizing the chemical potentials.
The maximum difference in chemical potentials between the traditional mixture (10 wt.% CBlack + 10 wt.% PVDF + 80 wt.% LFP) and the same mixture doctor bladed on the aluminum current collector, is ΔV ≈ 0.47 V = 0.12—(−0.35) V. The Al electrical contact to the 10 wt.% CBlack + 10 wt.% PVDF + 80 wt.% LFP mixture brings the OCV of the Li//LFP cell from 3.17 to 2.70 V vs Li (figure 5(c)). Depositing on the Cu would have a smaller impact, as shown in figure 5(f).
At C/3 cycling rates, the Li//LFP cell with liquid electrolyte charges by forming Li0.25FePO4; the first point is 3.40 V, but the subsequent evolution is at 3.49 V. The plateau corresponds to (1−x) LiFePO4 + x Li0.25FePO4. The Li-rich phase transforms into a Li-poor phase at 3.49 V (figure 5(g)). To 100% Li0.25FePO4 will correspond a maximum capacity of 124 mAh g−1, as calculated in figure 3(e).
If, however, the cell is a bilayer pack with LPSCl + LFP pressed together as described in SI, Methods, the Li/LPSCl/LFP cell does not perform well even at C/50, where the mobility of Li+ is still conditioned (figure 5(h)). The first charging potential is again 3.40 V like in figure 5(g). Still, the system advances to 3.57 V, which is the limit voltage for forming a plateau even with the metastable Li-poor Li0.25FePO4 (figure 3(e)). The all-solid-state cell cycles as a capacitor; all phenomena are surface-like, and the capacity reduces to <1 mAh g−1.
3. Conclusions
The primary purpose of this study was to intrinsically understand the behavior of the cathodes using the LFP as a case study, underlining the role of each constituent with experimental data supported by theoretical simulations provided by DFT and electrical transport analysis. With the simulated surfaces of the phases involved, the total local potentials leading to the workfunctions were simulated; with the bulk crystal structures, by simulating the DOS and carrier density analysis, it was possible to predict the materials' behaviors when electrically insulated and within the liquid and all-solid-state Li//LFP battery cells. The calculated parameters were all supported by the experimental data provided by SKP and electrochemical performance.
It was found that LFP is a semiconductor with a small band gap. It cannot equalize its chemical potentials with the other cathode or cell's materials if no carbon is present, and it is extremely sensitive to moisture. LFP shows a chemical potential that is very similar to Cblack and Cu.
A relatively small amount of carbon (3 wt.%) is enough to equalize the chemical potentials of the mixtures with the Cu electrode, whether the mixture corresponds to the traditional cathode or the solid electrolyte cathode mixture. Carbon (graphite) can have both a cathode and anode-like role.
The PVDF was found to be just a binder-like material with little to no capacity to polarize within the cathode.
Aluminum and Lithium 'working' chemical potentials deviate from the experimental and calculated workfunctions, as previously known, but coincide with chemical potentials of the highest DOS.
Current collectors also contribute to the overall chemical potential of the cathode.
The optimum limits of charging and discharging a Li//LFP were found to be 4.12 and 2.80 V. Above and below these values, both phases LFP and FePO4 start to oxidize and reduce, deteriorating and parasitic/uncontrolled reactions will take place, forming interfaces and forming interphases, which directly affect the performance of the battery.
The theoretical and metastable capacity of the Li//LFP cell, from 3.42 to 3.57 V, may be calculated using charge carriers considerations, assuming that charge and discharge happen in a range where the phases are stable and their number of charge carriers constant.
The number of LFP charge carriers at equilibrium is 8.50 × 1020 cm−3, which is the optimal number of charge carriers in thermoelectrics and superconductors, bringing together energy harvesting and storage devices and contributing to a unifying theory around these devices.
In summary, all battery target numbers may be extracted from the electronic structure and transport, independently of electrodes and electrolytes, paving the way for a comprehensive understanding of landmarks in battery science.
Acknowledgments
This research was funded by PULSELiON—PUlsed Laser depoSition tEchnology for soLid State battery manufacturIng supported by digitalization funded by Horizon Europe, EU, n. 101069686 . This work is also result of CVB agenda—Portuguese batteries value chain, nr. C644864613-00000003, financed by the Recovery and Resilience Plan (PRR) and by European Union—NextGeneration EU and the Portuguese Foundation for Science and Technology FCT UIDP/50022/2020 Emerging Technologies–LAETA, and PTDC/QUI-ELT/2593/2021 'Redox-active Metal-Organic Frameworks as Electrode Materials for Lithium-Ion Batteries' projects. B A M thanks the Fundação para a Ciência e Tecnologia (FCT), Portugal, for the Ph.D. Grant 2023.01813.BD. M H B acknowledges Professor John B Goodenough's endowment to the MatER–Materials for Energy Research lab, FEUP.
Data availability statement
The data cannot be made publicly available upon publication because the cost of preparing, depositing and hosting the data would be prohibitive within the terms of this research project. The data that support the findings of this study are available upon reasonable request from the authors.
Contributions
Cell fabrication and experiments: ANG, BAM, BMG; original draft: BAM, BMG, MHB; acquisition of the financial support for the project leading to this publication: RMS, MHB; conceptualization, formal analysis, simulations, and supervision: MHB; review, and editing: All. All authors have read and agreed to the published version of the manuscript.
Supplementary data (6.9 MB DOCX)