
Abstract
Silicon-containing steel forms an oxide layer within the scale during hot rolling. This layer comprising oxides such as Fe2SiO4 and FeO adversely affect the surface quality of steel owing to strong adhesion between Fe2SiO4 and the steel substrate. A method to melt this silicon-containing oxide layer through the oxidation reaction of iron plates by applying SiO2 powder to the surface is proposed. The aim of this study is to understand the reaction mechanism and identify the optimal conditions for this process. Application of SiO2 coating to the surface of an iron plate at 6.67 × 10–4 g·mm−2 and maintaining the plate at 1423 K, just below the eutectic temperature of Fe2SiO4–FeO system (1450 K), cause the surface oxide scale to melt when exposed to oxidation in an atmospheric environment. Notably, reducing the electric furnace temperature hinders the melting process, whereas excessive application of SiO2 hampers the formation of sufficient liquid phase. On the surface of the iron plate, primarily FeO and Fe2SiO4 are formed. Heat generation is mainly due to the formation of FeO, which leads to the rapid generation of Fe2SiO4. Eutectic reactions within SiO2–Fe2SiO4 or FeO–Fe2SiO4 systems drive the formation of the liquid phase. Once the liquid phase was established, rapid diffusion of iron ions within it resulted in rapid iron oxidation and additional heat generation. Ultimately, a liquid phase in equilibrium with FeO is formed across the entire surface of the iron plate.
Similar content being viewed by others

1 Introduction
Most commercial steel products comprise Fe-Si alloys as fundamental components owing to their strength enhancing characteristics. For example, high-tensile steel sheets used to build automobile chassis contain Si because its addition increases the strength of steel without degrading product ductility [1]. Steel sheets for automotive and other applications are manufactured using a hot-rolling process during which high-temperature oxidation results in formation of a layer, known as an “oxide scale,” on the surface of the steel plates. The presence of oxide scales could potentially induce surface defects, which significantly reduce the quality of steel products. This issue is particularly more profound in Si-containing steels wherein an oxide layer comprising Fe2SiO4, FeO, and pores is formed within the scale [2,3,4,5]. Fe2SiO4 is an intermediate compound formed through a reaction involving SiO2, which is introduced by the oxidation of Si in steel, and FeO formed by the oxidation of iron. There are several reports on the mechanism of oxide-scale formation when Fe-containing Si gets oxidized. For commercial Si-killed steel, Fukagawa et al. [6] reported that the adhesion strength between Fe2SiO4 and steel remains high below the melting point of Fe2SiO4 thereby making it harder to remove the oxide scale. Consequently, descaling (removal of the oxide scale) of Si-containing steel is typically done at temperatures at which the oxide scale melts. The melting point of Fe2SiO4 is 1490 K and the pseudo-binary phase diagram of FeO and Fe2SiO4 exhibit the eutectic forms [7, 8]. Because the eutectic temperature is 1450 K [8], the steel plate is typically heated above 1450 K in a heating furnace before hot-rolling.
However, Okada et al. [9] investigated a primary scale of high-Si steel (0.5 % Si) in simulated conditions of an actual heating furnace at 1493 K. They reported that even after descaling an oxide scale (mainly FeO) with thickness of 40–180 μm remained, and the steel is rolled with this scale still in place. Moreover, when maintained at 1473 K, which is just above the eutectic temperature, intricately shaped Fe2SiO4 (fayalite) layer is formed at the scale–steel interface thereby rendering descaling even more difficult. Therefore, a technique is needed to remove the aforementioned oxide scale from the steel plates from which the primary scale has already been removed.
Notably, the reactions responsible for forming FeO and Fe2SiO4, as shown in Eqs. 1 and 2, are exothermic. The following equations show example values at 1400 K [7, 10].
where ΔH(T) represents the enthalpy change of the reaction at temperature T. Based on the exothermic reaction patterns associated with the formation of FeO and Fe2SiO4, occurrence of localized heating of the steel surface is predicable. In such instances, the surface temperature may surpass the eutectic temperatures of Fe2SiO4–FeO system even when the overall steel temperature remains below 1450 K. The enthalpy change for the production of 1 mol of Fe2SiO4 from Fe(γ), O2(g), and SiO2 (cristobalite) can be determined as ΔH (1400 K) = − 568.305 kJ·mol−1, exceeding the enthalpy change for the melting of Fe2SiO4, ΔHmelt = 92.174 kJ·mol−1 [10]. This implies that it is possible to melt oxide scale containing Fe2SiO4 using surface oxidation reactions even when the steel plate temperature is below 1450 K. Fukumoto et al. [11] conducted a study on the high-temperature oxidation of 0–1.5 % Si–Fe in the 1323–1473 K temperature range. Notably, oxidation at 1373 K for 0.4 % Si and at 1423 K for 0.5 % Si and 1 % Si resulted in temperature elevation due to the oxidation reaction along with the formation of a liquid phase simultaneously.
When Si in steel undergoes oxidation, it is consumed, and the quantity of the resulting liquid phase depends on the Si concentration. By introducing SiO2 in powder form by external feed to the surface of the steel plate, it is feasible to generate a sufficient quantity of liquid phase, including Fe2SiO4, without exhausting the Si within the steel. Therefore, as part of a foundational investigation, this study aims to confirm the melting of an oxide layer containing Si through the oxidation reaction of a steel plate of which surface is coated with SiO2 powder and to elucidate the underlying mechanisms and required conditions for this process to be accomplished.
2 Experimental Procedure
Square iron plates, each 15 × 15 mm in size, with the thickness of 3 mm (99.99 % mass purity) were used. The plate samples were sequentially polished with #600, #1000, and #2000 grit abrasive papers, followed by ultrasonic cleaning with ethanol. Subsequently, three R-type thermocouples (φ = 0.3 mm) were spot welded to the upper and lower surfaces and edge of the one side of each test sample. Finally, approximately 0.15 g of SiO2 (quartz) powder (equivalent to 6.67 × 10–4 g·mm−2) was applied to the upper surface of each sample with a spatula, and a sufficient amount of ethanol was added to distribute the SiO2 powder evenly. The applied ethanol evaporated naturally at room temperature, remaining SiO2 powder layer with a uniform thickness.
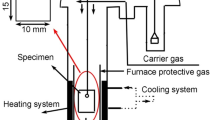
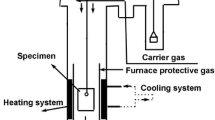
The test samples were positioned in a uniform heating zone within a horizontal electric furnace, as shown in Fig. 1(a). The inner diameter of the reaction tube was 42 mm. Figure 1(b) shows a schematic of the sample holder. The samples were positioned on alumina blocks, each with a thickness of 3 mm, to enable the diffusion of ambient gas to the lower surface of the sample. Because the φ0.3-mm thermocouples spot welded to the sample surface were delicate, φ0.5-mm R-type thermocouples were utilized for areas outside the measurement zone, at position F in Fig. 1(a). The furnace was initially heated to the experimental temperature (1373, 1423, or 1473 K) under nitrogen atmosphere with a flow rate of 200 ml·min−1. To prevent oxidation of the sample with residual oxygen in the nitrogen as the temperature rises in the furnace, the nitrogen gas was deoxygenated using magnesium at 773 K before injecting into the reaction furnace. Subsequently, the sample was oxidized by changing nitrogen gas to air gas with a flow rate of 400 ml·min−1, which was maintained for 30 min. The surface temperature of the samples was measured every 10 s during oxidation using spot-welded R-type thermocouples. After oxidation, the samples were removed from the furnace and allowed to cool. Comparative experiments were conducted with and without SiO2 powder coating. Moreover, the additional experiments were performed at 1423 K with two variations:

Schematic illustration of experimental setup: (a) apparatus and (b) sample holder
-
(i)
By applying 0.3 g of SiO2 powder for each sample, equivalent to 13.1 g·mm−2.
-
(ii)
By applying 0.15 g of SiO2 powder for each sample and removing the sample from the furnace 190 s after oxidation initiation.
Table 1 summarizes the sample designations, experimental conditions: holding temperature, and amount of SiO2 powder added. After the experiment, phase identification was performed using X-ray diffraction (XRD). Microstructural analyses were conducted by field-emission scanning electron microscopy (FE-SEM). Energy-dispersive X-ray spectroscopy (SEM) was used to identify the existing phases.
3 Results
3.1 Sample Photographs
Figure 2 shows the photographs of the upper and lower surfaces of the samples after the experiment. When oxidation was conducted in air without application of the SiO2 powder, a uniform oxide scale was observed on both the upper and lower surfaces, except for the areas on the lower surface that were in contact with the base of the alumina block. At 1473 K, the lower surface of the SiO2-free sample could not be photographed because the sample got adhered to the alumina-block sample holder. For sample S15-1373 (with SiO2 powder applied and held at 1373 K), a notable quantity of SiO2 powder remained on the upper surface. On the lower surface, the oxide scale is visible primarily in the upper portion of the photograph, resulting from scale detachment.

Appearance of upper and lower surfaces of each sample, where portions for oxide scale were not indicated because almost all samples were covered with oxide scale except upper surfaces of S15-1373 and S30-1423
Conversely, for the samples maintained at 1423 and 1473 K (samples S15-1423 and S15-1473), an uneven oxide scale could be observed on the upper surface. At 1423 K, the oxide scale detached from the lower surface when the sample is separated from the base. For S15-1473, an uneven oxide scale could be observed on both the upper and lower surfaces. For sample S30-1423, in which a greater quantity of SiO2 was applied, a SiO2 residue could be observed on the upper surface, whereas no uneven scale could be observed on the lower surface.
3.2 Result of Temperature Measurement
The temperature profiles after changing from nitrogen atmosphere to air atmosphere are shown in Fig. 3. Temperature measurements on the side edges of samples S15-1373 and S15-1473 were unsuccessful. For sample N-1373, the surface temperatures uniformly increased on the upper and lower surfaces as well as side edges after switching to air atmosphere for approximately 100 s. Subsequently, the temperature started to decrease; there were variations in the rate of temperature drop between the surfaces. Samples N-1423 and N-1473 exhibited temperature profiles similar to those of N-1373. The maximum temperature surpassed the eutectic temperature of Fe2SiO4–FeO system at 1450 K.

Change in surface temperature after switching atmosphere from N2 to air
Sample S15-1373 exhibited a temperature profile similar to that of sample N-1373, which does not contain SiO2 powder. However, for S15-1423 and S15-1473, temperature increase was observed approximately 100 s after the change in the oxidizing atmosphere. Subsequently, a rapid temperature fluctuation was observed that was distinct from the behavior of the samples without SiO2 powder (N-1373, N-1423, and N-1473) and S15-1373. Notably, these rapid temperature fluctuations were not limited to the upper surface alone, they could be observed on the side edges and lower surfaces as well, thus, implying occurrence of a melting reaction on the upper surface and flow of the resulting liquid phase to the side edges and lower surfaces.
For sample S30-1423, the upper surface temperature increased after oxidation initiation, following a pattern similar to the other results. The maximum temperature at the upper surface exceeded 1450 K, indicating that the oxide scale has melted. Furthermore, approximately 100 s after the initiation of oxidation, the temperature increase temporarily slowed, signifying the occurrence of an endothermic reaction associated with melting.
Based on the results shown in Figs. 2 and 3, the presence or absence of melting in each sample is summarized in Table 1. In addition, Table 1 shows the maximum temperature increase when switching to air atmosphere. For the samples without SiO2 powder (N-1373, N-1423, and N-1473), the maximum temperature increase during the oxidation process ranged from approximately 55 K to 70 K. These values did not display noticeable variability, and no clear correlation was observed between the surface temperature, holding temperature, and maximum temperature increase. For sample S15-1373, the temperature increase on the upper and lower surfaces remained at 58 and 57 K, respectively, which is quite contrary to that of samples S15-1423 and S15-1473 that were also subjected to SiO2 application. Samples S15-1423 and S15-1473 exhibited considerably; greater temperature increases than samples N-1373 and N-1473. Particularly, the temperature increased by as much as 368 K on the side edges of S15-1423. Temperature increase of sample S30-1423 was similar to that of sample N-1423.
3.3 XRD Results
The XRD results for the phase identification of the oxide scales in the post-experiment samples are displayed in Fig. 4, illustrating the results for samples N-1423 and S15-1423. The identified phases are listed in Table 2. When SiO2 powder was not applied, the detected phases included FeO, Fe3O4, and Fe2O3, whereas Fe2SiO4 was also detected when SiO2 powder was applied to these phases. Notably, although XRD did not identify Fe2SiO4 in sample S15-1373, it was found within the oxide scale through SEM–EDS analysis, as discussed later.

X-ray diffraction patterns of samples for (a) N-1423 and (b) S15-1423
3.4 Analysis Results by FE-SEM
At 1423 K, addition of 0.15 g of SiO2 powder resulted in melting of the oxide scale, whereas the same amount of SiO2 powder added at 1373 K did not melt. Even an increased amount, i.e., 0.3 g, at the same temperature did not show dramatic temperature change at the lower surface. To clarify this difference, cross-sectional observations of the samples were obtained. The results for each sample are shown in Figs. 5, 6, and 7.
Figure 5(a) illustrates the cross section of sample S15-1423, especially, the cross sections of the upper surface of sample. This S15-1423 sample displayed the melting of the oxide scale from the appearance. A 1.4-mm-thick oxide scale was observed on the upper surface, comprising layers of FeO/Fe2SiO4, FeO, and Fe3O4 originating from the iron side. The FeO/Fe2SiO4 layer occupied one half of the scale thickness. The bright areas correspond to FeO and exhibit a circular shape with diameter equal to several tens of micrometers. When magnified (as shown in Fig. 5(c) and (d)), it is evident that Fe2SiO4 and FeO are lamellar. This suggests that FeO and Fe2SiO4 precipitate from the molten material during cooling, forming a relatively large FeO layer in equilibrium with the molten phase. A scale phase approximately 1.5 mm thick on the lower surface displayed a layer structure similar to that of the upper surface. Interestingly, even though SiO2 was not applied to the lower surface, Fe2SiO4 was detected thereby indicating the flow of the molten material to the lower surface.

FE-SEM cross-sectional image of top surface of sample S15-1423: (a) upper surface of the sample including oxide scale and iron plate, (b), (c), and (d) magnified images for the parts as indicated in (a)
A cross-sectional image of the sample S15-1373 is shown in Fig. 6. A scale layer of approximately 0.16 mm thickness was observed on the upper surface, comprising layers of FeO + Fe2SiO4 and FeO + SiO2 originating from the iron side. This indicates that FeO and SiO2 reacted within the scale layer on the iron side, forming Fe2SiO4. The growth of Fe2SiO4 progressed in a layered manner from the Fe side. On the lower surface, a scale phase of approximately 0.62 mm thick consisting of FeO, Fe3O4, and Fe2O3 layers from the iron side was visible. The thickness ratio of each phase in the scale layer was FeO: Fe3O4:Fe2O3 = 94:5:1; this closely matches what has been reported by Mrowec et al. [12]. The thickness of the bottom surface is greater than that of the oxide scale on the upper surface. This can be attributed to the formation of Fe2SiO4 on the upper surface, which hinders the diffusion of Fe ions [13, 14].

BEI for cross section of sample S15-1373
Figure 7 shows the cross-sectional image of the sample S30-1423. Unreacted SiO2 powder remains on the surface. Meanwhile, the oxide scale on the iron side consists of FeO and Fe2SiO4, and this microstructure is similar to that of S15-1423. From the temperature profile, i.e., Figure 3(g), the maximum temperature of the sample S30-1423 was over 1450 K, suggesting that the scale was melted. The maximum temperature was lower than that for the sample S15-1423. The thickness of oxide scale of the sample S30-1423 is approximately 400 μm, which is thinner than that for the sample S15-1423. Thus, the presence of a thick SiO2 powder layer inhibited the supply of oxygen gas to the scale surface, resulting in slower progress of oxide scale melting and less heat generation than in the sample S15-1423.

BEI for cross section of sample S30-1423
Figure 8 displays the temperature change, sample appearance, and backscattered electron image (BEI) for a sample that began oxidation under the same conditions as S15-1423 and was removed after 190 s into the process. The temperature change shown in Fig. 8(a) reveals that the upper surface of the sample reached 1467 K after 100 s of oxidation initiation, decreased to 1458 K, and then began to increase again before removal. This decrease in temperature can be attributed to the endothermic effect of melting.

(a) Temperature change for cross section of sample oxidized under same condition as S15-1423 for 190 s, (b) photos of sample, and cross-sectional BEIs for portions (c) SiO2 powders on the surface, and (d) the magnified image and (e) no SiO2 powder on the surface and (f) magnified image
Figure 8(c) shows the analysis results for the portion of the SiO2 powder remaining on the upper surface, indicating the presence of FeO (bright gray) in the upper section. The oxide scale close to the iron substrate appeared to have melted, forming FeO/Fe2SiO4. The majority of the oxide consisted of Fe2SiO4 (gray) and unreacted SiO2 (dark gray). Upon further magnification (Fig. 8(d)), it became evident that there were laminar regions of darker and lighter gray. Although analyzing these phases via SEM–EDS was challenging because of their small size, this region likely contained SiO2 and Fe2SiO4 in laminar form. Therefore, this part is believed to have been a molten phase before the sample was removed from the furnace, and SiO2 and Fe2SiO4 were separated through a eutectic reaction during cooling.
On the other hand, Fig. 8(e) displays the cross-sectional SEM of the portion without unreacted SiO2 powder on the upper surface, revealing the presence of SiO2 internally. Figure 8(f) depicts the zoom-in image of the central part of Fig. 8(e), primarily comprising SiO2 and Fe2SiO4. In addition, there is also a bright layer that can be considered as FeO. Owing to the laminar arrangement of FeO and Fe2SiO4, it is likely that this region has already melted at the interface on the iron side.
4 Discussion
4.1 Effect of Application of SiO2 Powder
Based on the previous results, it was observed that even when the temperature of the iron plate was maintained at 1423 K, which is below the eutectic temperature of FeO–Fe2SiO4 system (1450 K), application of SiO2 powder resulted in melting of the oxide scale. To better understand the influence of SiO2 powder application, a comparison was made between the cases with and without SiO2 at different holding temperatures, and the results are presented in Fig. 9.

Comparison of temperature change for experiments at the same temperature: (a) upper surface at 1373 K, (b) bottom surface at 1373 K, (c) upper surface at 1423 K, (d) bottom surface at 1423 K, (e) upper surface at 1473 K, and (f) bottom surface at 1473 K
Figure 9(a) and (b) illustrates a comparison at 1373 K, where the initial temperature increase was consistent regardless of the presence of SiO2. However, sample N-1373, which did not have SiO2 applied, exhibited a temperature increase approximately 10 K higher than that of sample S15-1373 on both the upper and lower surfaces. The surface temperature did not exceed 1450 K, even for the iron plate without SiO2 powder, during the oxidation reaction. As shown in Fig. 6, FeO and Fe2SiO4 were generated on the surface of the iron plate. The heat generation on the upper surface of the iron plate with SiO2 powder application can be attributed to the simultaneous occurrence of two different reactions: FeO and Fe2SiO4. However, the formation of FeO was slower, especially in the initial period, resulting in a lower temperature rise compared with the case without SiO2 powder application. Two factors contributed to the slow growth of FeO: first, the reduced supply of oxygen to the FeO surface owing to SiO2 powder application; second, FeO growth is hindered by the presence of Fe2SiO4, as FeO and Fe2SiO4 coexist.
Consequent to switching to air atmosphere, it was observed at 1423 K that the temperature rise was consistent for cases with and without SiO2 powder application for approximately 100 s, as depicted in Fig. 9(c) and (d). Subsequently, the temperature decreased in the case without SiO2 powder application. In contrast, in the case subjected to SiO2 powder application, both the upper and lower surfaces exhibited a rapid increase in temperature, followed by a sharp decrease on the upper surface and a gradual decrease on the lower surface. The rapid temperature increase on the upper surface indicates the generation of molten material and an associated increase in the oxidation rate. Previous reports have suggested that the generation of molten material during the oxidation of iron-containing silicon results in an accelerated oxidation rate. The subsequent temperature decrease on the upper surface of samples S15-1423 can also be attributed to increase in the amount of molten material and the associated endothermic effect. However, it is unlikely to drop to 1320 K, which is more than 100 K below the furnace temperature. The generated molten material may affect the measured thermoelectric potential. In other words, the molten material might have entered between the two wires of the thermocouple. Since the FeO-SiO2 melt has electrical conductivity (e.g., 100–400 Ω−1 m−1 at 1526 K [15]), the wires are not insulated from each other, which may have reduced the electromotive force. At 1473 K, the initial temperature rise was faster when SiO2 was applied, indicating rapid heat generation at sufficiently high temperatures: FeO was generated, which quickly reacted with SiO2 to produce Fe2SiO4.
4.2 The Mechanism of Oxide Scale Growth on Iron Plates with SiO2 Powder Application
In this section, we discuss the growth mechanism of oxide scale for the sample with SiO2 powder application maintained at 1423 K, where the formation of a liquid phase was confirmed. Figure 8 indicates that the initial liquid phase generated at the beginning of the oxidation-melting reaction was within the SiO2-Fe2SiO4 and FeO-Fe2SiO4 systems. Furthermore, as demonstrated in Fig. 5, the final liquid phase can be considered to be in equilibrium with FeO. These findings can be explained using the SiO2–FeO binary phase diagram depicted in Fig. 10, which was calculated using FactSage [16]. Unlike previous studies, FactSage calculated that the eutectic temperature of FeO–Fe2SiO4 system was 1466 K and that of SiO2–Fe2SiO4 system was 1462 K.

Schematic diagram of the reaction pathway on SiO2–FeO phase diagram
The liquid phase formed at the outset of oxidation is expected to be at the eutectic composition of the SiO2–Fe2SiO4 system (point A) and FeO–Fe2SiO4 system (point B). The endothermic reaction that occurs during the generation of the liquid phase results in decrease in temperature (Fig. 8(a)). However, once the molten material is formed, the diffusion of iron ions into the liquid phase is rapid and accelerates the oxidation process [17]. Consequently, the temperature increases owing to the exothermic reactions associated with iron oxidation.
4.3 Estimation of Heat Generation Change
The reactions described in Eqs. 1 and 2 occurred simultaneously in the sample with SiO2 powder application. Moreover, both reactions were exothermic. To form 1 mol of Fe2SiO4, 2 mol of Fe was consumed, and 2 mol of FeO was generated through the following reaction:
The heat generation for the formation of 1 mol of Fe2SiO4 at 1423 K was 538 kJ for the FeO formation step in Eq. 3 and 29 kJ for the formation of Fe2SiO4 in Eq. 4, indicating that the FeO formation reaction contributed to 95 % of the total heat generation. Formation of Fe2SiO4 was disregarded in this context because it was not present during the initial oxidation stage.
Using FactSage, the enthalpy changes in the eutectic reactions (points A and B in Fig. 10) were determined to be 38 and 37 kJ·mol−1, respectively.
These enthalpy changes are significantly lower than the enthalpy released during the formation of FeO (538 kJ·mol−1), as indicated in Eq. 3. This confirms that most of the heat generated during the process can be attributed to the formation of FeO. The FeO that was generated was incorporated into the molten phase subsequently thereby increasing the concentration of FeO in the liquid phase and following a path along the liquidus line. Consequently, the temperature continued to increase. The FeO concentration in the liquid phase decreased upon cooling, leading to the precipitation of solid-phase FeO.
The time evolution of heat generation can be estimated by considering the thickness of the oxide scale and the initial temperature rise. Based on the results for sample N-1423, thickness of the oxide scale formed when iron was oxidized at 1423 K for 1800s was 0.9 mm. Assuming a parabolic growth model, the thickness of the oxide scale can be described using the formula: l = 0.0212√t, where l is the thickness of the oxide scale (mm) and t is time (s).
The heat generation rate was estimated by assuming that all the generated oxides were FeO, as shown in Fig. 11, using the thermodynamic data [10]. The heat generation rate at 1 s after the start of the reaction was 0.014 J·mm−2s. It decreased to 1/20 of this rate 100 s after the reaction began. During the 1800s of the reaction, 24 % of the total heat generation occurred in the initial 100 s. Therefore, increasing the surface temperature above the eutectic temperature of SiO2–Fe2SiO4 system is essential in the early stages when the heat generation rate is high. This can be achieved by increasing the oxygen concentration during the initial oxidation phase to expedite the formation of FeO.

Estimated heat generation by the oxidation reaction of iron as a function of time
This study reveals that the application of SiO2 powder allows the melting of the oxide scale on iron plates at a minimum temperature that is between 1373 K and 1423 K. To determine the exact temperature, start by identifying the furnace temperature at which the surface temperature of the iron plate without the SiO2 coating exceeded 1450 K during oxidation was used. Subsequently, the SiO2 quantity was adjusted to determine the melting temperature.
5 Conclusions
In this study, we applied SiO2 powder to the surface of iron plates to confirm the melting of the oxide layer containing Si through the oxidation reaction of the iron plate and elucidated its mechanism and conditions. The results are summarized as follows:
When SiO2 is applied to the surface at a rate of 6.67 × 10–4 g·mm−2, and the iron plate is maintained at a temperature of 1423 K, which is lower than the eutectic temperature of FeO–Fe2SiO4 system (1450 K), the surface oxide scale melts. At a lower electric furnace temperature, no melting occurs. Moreover, with an excessive SiO2 quantity, an insufficient amount of the liquid phase is generated.
On the surface of the iron plate, primarily FeO and Fe2SiO4 are formed. Heat generation owing to the reaction primarily results from the formation of FeO, which leads to rapid generation of Fe2SiO4.
The liquid phase is generated through eutectic reactions in SiO2–Fe2SiO4 or FeO–Fe2SiO4 systems.
Once the liquid phase is formed, the rapid diffusion of iron ions in the liquid phase leads to rapid iron oxidation and additional heat generation. Ultimately, the composition of the liquid phase reached equilibrium with FeO.
Data Availability
No datasets were generated or analyzed during the current study.
References
C.G. Lee, S.J. Kim, C.S. Oh, S. Lee, ISIJ Int. 42, 1162 (2002). https://doi.org/10.2355/isijinternational.42.1162
R.C. Logani, W.W. Smeltzer, Oxid. Met. 3, 15 (1971). https://doi.org/10.1007/BF00609922
X. Liu, G. Cao, Y. He, T. Jia, Z. Liu, J. Iron. Steel Res. Int. 20, 73 (2013). https://doi.org/10.1016/S1006-706X(13)60199-5
A. Alaoui Mouayd, A. Koltsov, E. Sutter, B. Tribollet, Mater. Chem. Phys. 143, 996 (2014). https://doi.org/10.1016/j.matchemphys.2013.10.037
E.J. Song, D.W. Suh, H.K.D.H. Bhadeshia, Ironmak. Steelmak. 39, 599 (2012). https://doi.org/10.1179/1743281212Y.0000000007
T. Fukagawa, H. Okada, Y. Maehara, ISIJ Int. 34, 906 (1994). https://doi.org/10.1179/1743281212Y.0000000007
I. Barin, Thermochemical Data of Pure Substances (Wiley, New York, 1995), p.713. https://doi.org/10.1002/9783527619825
A. Muan, Am. J. Sci. 256, 171 (1958). https://doi.org/10.2475/ajs.256.3.171
H. Okada, T. Fukagawa, H. Ishihara, A. Okamoto, M. Azuma, Y. Matsuda, Tetsu-to-Hagane 80, 849 (1994). https://doi.org/10.2355/tetsutohagane1955.80.11_849
I. Barin, Thermochemical Data of Pure Substances (Wiley, New York, 1995), p.700. https://doi.org/10.1002/9783527619825
M. Fukumoto, S. Hayashi, S. Maeda, T. Narita, Testu-to-Hagane 85, 878 (1999). https://doi.org/10.2355/tetsutohagane1955.85.12_878
S. Mrowec, A. Podgorecka, J. Mater. Sci. 22, 4181 (1987). https://doi.org/10.1007/BF01132007
C.W. Tuck, Corros. Sci. 5, 631 (1965). https://doi.org/10.1016/S0010-938X(65)90189-7
S. Taniguchi, K. Yamamoto, D. Megumi, T. Shibata, Mater. Sci. Eng. A 308, 250 (2001). https://doi.org/10.1016/S0921-5093(00)01977-8
Veerein Deutscher Eisenhuttenleute(VDEh), Slag ATLAS 2nd Edition (Verlag Stahleisen GmbH, 1995), p. 564
C.W. Bale, P. Chartrand, S.A. Degterov, G. Eriksson, K. Hack, R. Ben Mahfoud, J. Melançon, A.D. Pelton, S. Petersen, Calphad 26, 189 (2002). https://doi.org/10.1179/1743281212Y.0000000007
D.P. Agarwal, D.R. Gaskell, Metall. Trans. B 6, 263 (1975). https://doi.org/10.1007/BF02913568
Acknowledgements
The authors thank Mr. Shintaro Ono at Shibaura Institute of Technology for conducting part of the SEM-EDS analysis. The XRD analysis was conducted at the Materials Analysis Division, Open Facility Center of the Tokyo Institute of Technology. We would like to thank Editage (www.editage.com) for the English language editing.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
R.E conceived and planned the experiments. K.M and S.H. performed the experiments and analysis. L.M. and T.W. conducted the calculation in discussion. M.H. and M.S. helped supervise K.M. M.U. and H.T. discussed the results. M.S. and T. W. decided on the experimental conditions. R.E. wrote the main manuscript text. All authors discussed the results and contributed to the final manuscript.
Corresponding author
Ethics declarations
Competing interest
The authors declare no competing interests.
Ethical Approval
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Selected Papers of the 22nd European Conference on Thermophysical Properties.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Endo, R., Mitsui, K., Hashiyama, S. et al. Potential Utilization of Iron Oxidation Reaction Heat for the Generation of SiO2–FeO System Melts. Int J Thermophys 45, 44 (2024). https://doi.org/10.1007/s10765-024-03338-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10765-024-03338-y