
Abstract
Four species of Saussurea, namely S. involucrata, S. orgaadayi, S. bogedaensis, and S. dorogostaiskii, are known as the “snow lotus,” which are used as traditional medicines in China (Xinjiang), Kyrgyzstan, Mongolia, and Russia (Southern Siberia). These species are threatened globally, because of illegal harvesting and climate change. Furthermore, the taxonomic classification and identification of these threatened species remain unclear owing to limited research. The misidentification of medicinal species can sometimes be harmful to health. Therefore, the phylogenetic and genomic features of these species need to be confirmed. In this study, we sequenced five complete chloroplast genomes and seven nuclear ITS regions of four snow lotus species and other Saussurea species. We further explored their genetic variety, selective pressure at the sequence level, and phylogenetic relationships using the chloroplast genome, nuclear partial DNA sequences, and morphological features. Plastome of the snow lotus species has a conserved structure and gene content similar to most Saussurea species. Two intergenic regions (ndhJ–ndhK and ndhD-psaC) show significantly high diversity among chloroplast regions. Thus, ITS and these markers are suitable for identifying snow lotus species. In addition, we characterized 43 simple sequence repeats that may be useful in future population genetic studies. Analysis of the selection signatures identified three genes (rpoA, ndhB, and ycf2) that underwent positive selection. These genes may play important roles in the adaptation of the snow lotus species to alpine environments. S. dorogostaiskii is close to S. baicalensis and exhibits slightly different adaptation from others. The taxonomic position of the snow lotus species, confirmed by morphological and molecular evidence, is as follows: (i) S. involucrata has been excluded from the Mongolian flora due to misidentification as S. orgaadayi or S. bogedaensis for a long time; (ii) S. dorogostaiskii belongs to section Pycnocephala subgenus Saussurea, whereas other the snow lotus species belong to section Amphilaena subgenus Amphilaena; and (iii) S. krasnoborovii is synonymous of S. dorogostaiskii. This study clarified the speciation and lineage diversification of the snow lotus species in Central Asia and Southern Siberia.
Similar content being viewed by others


Introduction
Saussurea DC. is one of the most diverse genera in the Asteraceae family, with approximately 460–490 herbaceous species that are mainly prevalent in the alpine habitats of the Himalayas and temperate regions of Asia (Chen 2015; Raab-Straube 2017; Xu et al. 2019). This genus merged in the early-middle Miocene within the Hengduan Mountains (South Asia) and dispersed northward due to the Himalayan uplift and adjacent mountain range formation (Chen 2015). As a result, 150 species are distributed in Central Asia, and 100 species are distributed in Eastern Asia (Lipschitz 1979; Wang et al. 2009; Baasanmunkh et al. 2022). The Saussurea genus is divided into four subgenera, Saussurea DC., Eriocoryne (DC.) Hook. f., Amphilaena (Stschegl.) Lipsch., and Theodorea (Cass.) Lipsch. and approximately 15 sections, based on morphological characteristics (Wang et al. 2009; Shi and Raab-Straube 2011; Zhang et al. 2021a). Among them, Amphilaena is particularly rich in medicinal resources, and comprises 39 taxa (Chik et al. 2015; Raab-Straube 2017; Chen et al. 2019). This subgenus has a glabrous stem, leafy bracts, and solitary capitula, which have many adaptive traits from multiple origins and arose through convergent evolution (Zhang et al. 2021a). However, this characteristic is a well-known strategy for adaptation to high altitudes and occurs in other angiosperms from several families (Omori et al. 2000).
Misidentified, rare, medicinally important species belonging to the subgenus Amphilaena are distributed in Central Asia and Southern Siberia. Particularly, Saussurea bogedaensis Yu J.Wang & J.Chen, Saussurea involucrata (Kar. & Kir.) Sch.Bip., Saussurea orgaadayi Khanm. & Krasnob., and Saussurea dorogostaiskii Palib. (or Saussurea krasnoborovii S.V.Smirn) poorly investigated based on morphological and molecular evidence. For instance, S. orgaadayi and S. involucrata have been distributed in the western and central parts of Mongolia for a long time; however, the taxonomic circumscription of these two species is difficult. Recently, another similar species, S. bogedaensis, was discovered, which was previously known as S. involucrata or S. orgaadayi (Chen and Wang 2018; Baasanmunkh et al. 2020). Moreover, the taxonomic statuses of S. dorogostaiskii and S. krassnorobovii remain unclear (Smirnov 2004; Raab-Straube 2017; Smirnov et al. 2018). In traditional medicine, the abovementioned species are known as “snow lotus or vansemberuu” and are used to treat lung diseases (Nyambayar et al. 2011; Chik et al. 2015; Norris 2020). However, given these species are used for medicinal purposes, misidentification can be fatal to humans (Chik et al. 2015). Thus, there is an urgent need to identify possible substitutes for these species.
Phylogenetic relationships of the subgenus Amphilaena, based on the nuclear ribosomal DNA (nrDNA) (ITS) and plastid (matK, rbcL, trnK, trnH-psbA) barcoding markers, explored by Chen et al. (2019) showed that ITS and rbcL markers were well identified in all species. However, based on the chloroplast whole-genome (plastome) analysis of Saussurea, these chloroplast markers failed to resolve relationships across the genus (Zhang et al. 2019). Recently, some classifications and diversity patterns of Saussurea have been revised based on morphological and ecological traits and plastome (Xu et al. 2019; Zhang et al. 2019, 2021a, c). Chloroplasts are organelles that play an essential role in photosynthesis in green plants (Bruneau et al. 2007). They have a maternally inherited haploid genome and have the potential to significantly advance the resolution of evolutionary relationships among complex plant lineages (Ravi et al. 2008). They can be used to infer well-resolved phylogenetic relationships, even at the species level, and for species classification and population genetic studies (Jansen et al. 2007). Positive selection of chloroplast genes can also improve our knowledge of plants adapting to extreme environments in alpine regions (Bock et al. 2014). The application of divergent regions and simple sequence repeats (SSRs) as DNA barcodes can guarantee species identification (Shen et al. 2020). In addition to plastome data, nrDNA, which constitutes the catalytic core of ribosomes, is also widely used for phylogenetic studies in terrestrial plants (Rodnina et al. 2007). The 45S nrDNA units are composed of three subunits (18S, 5.8S, and 26S rDNAs) and two internal transcribed spacer regions (ITS1 and ITS2), while thousands of 45S units are tandemly repeated in the nuclear genome (Long and Dawid 1980). Owing to its structural advantages, this sequence is highly conserved, making it a useful resource for phylogenetic studies of terrestrial plants (Lagesen et al. 2007). Therefore, the plastome and nrDNA sequences and morphology features are necessary for identifying the snow lotus species.
This study aimed to characterize the plastome of snow lotus species in Central Asia and Southern Siberia, and identify genetically variable regions for the development of informative chloroplast DNA markers through comparisons with closely related species within the Amphilaena subgenus, to reconstruct the phylogenetic relationships among the snow lotus species. This study seeks to provide insight into the evolution of Saussurea species in Central Asia and Southern Siberia.
Materials and methods
Plant materials, DNA extraction, PCR amplification, and sequencing
In this study, fresh leaves from S. bogedaensis, S. orgaadayi, S. dorogostaiskii, and S. involucrata from the snow lotus group and Saussurea baicalensis (Adams) B.L.Rob. from Mongolia, Kyrgyzstan, and Russia, collected since 2016, were used (Table S1). We did not collect herbarium specimens of the snow lotus group from the wild because they are a globally threatened species. Moreover, we examined specimens from the herbarium of Komarov Botanical Institute RAS, Moscow University, Martin-Luther-Universität Halle-Wittenberg, Central Siberian Botanical Garden, Tomsk State University, and National Univesity of Mongolia, according to Thiers (2023). Detailed photographs of all species were taken by the authors during the field surveys. The distribution maps were created using ArcGIS (ESRI 2011) based on field collections and herbarium specimens.
Total genomic DNA was extracted from silica gel-dried leaf material using the CTAB method (Doyle and Doyle 1987). The nuclear ITS region (White et al. 1990) was used for the PCR amplification and Sanger sequencing. PCR amplification was performed using 100 ng/μl of the sample, as described in previously (Baasanmunkh et al. 2020). PCR products were sequenced in both directions by Macrogen (Seoul, Korea). Sequences were aligned using ClustalW (Thompson et al. 2003); BioEdit was used to set the default settings and perform manual adjustments (Hall et al. 2011). Ambiguous nucleotide bases were corrected using corresponding bases of sequences obtained using a reverse primer. We sequenced the plastome of one accession from each snow lotus species. The plastome library was prepared from the total genomic DNA using the TruSeq DNA Nano Kit, along with NextSeq 500 platform (Illumina, San Diego, CA, USA), following the manufacturer’s protocol. Trimmomatic v. 0.36 (Bolger et al. 2014) was used to remove adapter sequences and low-quality reads to reduce bias. The total number of bases and reads, GC content (%), and the Q20 (%) and Q30 (%) scores were calculated after filtering. A base quality plot generated using FastQC v. 0.11.5 (Antil et al. 2023) was used to check the overall quality of the data and shows the range of quality values for each cycle.
Plastome assembly/annotation
NOVOplasty v.4.1.0 was used to perform de novo assembly using various k-mers (Nicolas et al. 2017). The best k-mer was selected based on the assembly results, including the number of contigs, total length of contigs, and N50. The raw data reads were mapped to an assembly result to identify the insert size of the raw data and number of reads used in the assembly. The assembly and orientations were confirmed using BLAST tool of NCBI (https://www.ncbi.nlm.nih.gov/) and graphic views using Geneious Prime® 2023.2.1 (https://www.geneious.com; Biomatters Ltd.) and Geseq—annotation of organellar genomes (Tillich et al. 2017). The transfer RNAs (tRNAs) were identified using tRNA-scan-SE (Lowe and Chan 2016). A circular chloroplast genomic map was visualized using Chloroplot online software (https://irscope.shinyapps.io/Chloroplot/) (Zheng et al. 2020).
Plastome analysis
The pairwise whole plastome alignment was visualized using MAUVE v2.3.1 (Darling et al. 2010). The boundary shifts in the single copy (SC)/inverted repeat (IR) at four junctions of the plastomes were compared using IRscope (https://irscope.shinyapps.io/irapp/) (Amiryousefi et al. 2018). The mVISTA program (Frazer et al. 2004) in the Shuffle-LAGAN mode (Brudno et al. 2003) was used to detect species-specific genetic variation by comparing newly sequenced plastomes, with Saussurea obvallata (DC.) Sch.Bip. as a reference.
Repeat sequence analysis
We used REPuter to identify forward, reverse, palindromic, and complementary repeats with a minimum length of 20 bp, 90% identity, and a Hamming distance of 3 (Kurtz 2001). SSRs were detected using MISA (Beier et al. 2017), with the minimum number of repeat parameters set to 10, 5, 4, 3, 3, and 3 for mononucleotides, dinucleotides, trinucleotides, tetranucleotides, pentanucleotides, and hexanucleotides, respectively. Tandem repeats ≥ 20 bp were identified using the tandem repeat finder (Benson 1999) with a minimum alignment score of 50 and a maximum period size of 500; the identity of repeats was set to ≥ 90%.
Genome divergence
The five newly sequenced samples, S. involucrata, S. orgaadayi, S. bogedaensis, S. dorogostaiskii, and S. baicalensis, and four species from the NCBI database were used in this experiment. The plastomes was individually aligned using MAFFT ver. 7.388 (Katoh 2002). Sliding window analysis was used to calculate the nucleotide variability (Pi) among the plastomes using DnaSP v.6.11 (Rozas et al. 2017). The step size was set to be 200 bp, with a 600-bp window length. Protein-coding genes (coding regions), intergenic spacers, and introns (non-coding regions) of the plastomes were extracted separately to screen for polymorphic hotspots. The relative synonymous codon usage (RSCU) of the plastomes was analyzed using the MEGA11 software (Tamura et al. 2021). The codon usage distribution of snow lotus plastomes was visualized using the Heatmapper tool with average linkage clustering and Euclidean distance measurement methods (Babicki et al. 2016). An RSCU < 1.00 indicated a codon that was used less frequently than expected, whereas an RSCU > 1.00 indicated a codon that was used more frequently than expected. Selective pressure was analyzed for consensus exons of the protein-coding genes in the snow lotus species using KaKs_Calculator 2.0 (Wang et al. 2010) with calculation mode NY and genetic code 11 (bacterial and plant plastid code), using S. obvallata as a reference. Genes with a Ka/Ks > 1 ratio were considered to be under positive selection, and genes with a Ka/Ks ratio < 1 were considered to be under purifying selection.
Phylogenetic analysis
The nrDNA ITS region and plastome sequences of the Saussurea species included in this study were used to determine the phylogenetic position of the snow lotus species within the Saussurea genus. Plastome alignment datasets were filtered to remove ambiguously aligned regions using GBlocks ver. 0.91.1 (Talavera and Castresana 2007). The best-fitting model for nucleotide substitutions was determined using the Akaike information criterion in jModelTest v2.1.10 (Darriba et al. 2012), and the GTR + I + G model was selected for maximum likelihood (ML) analysis. The GTR + G model was selected for the Bayesion inference (BI) analysis. The maximum parsimony (MP) analysis was conducted using PAUP* v4.0b10 (Swofford and Documentation 1989); the searches included 1000 random addition replicates and tree bisection and re-connection branch swapping using the MulTrees option. ML analysis was performed using RaxML v. 8.0.5 (Stamatakis 2014), with 1000 bootstrap replicates. BI analysis was performed using MrBayes 3.2.2 (Ronquist et al. 2012), with two independent runs of four simultaneous chains, executed for 5,000,000 generations using the Markov chain Monte Carlo algorithm. Trees were sampled every 5000 generations, and the first 25% were discarded as burn-in. The trees were determined using a 50% majority-rule consensus to estimate posterior probabilities (PPs). The reconstructed trees were visualized using FigTree v.1.4.2 (Rambaut 2012).
Results
Comparison of the morphological characteristics of the snow lotus species
Morphological differences among the snow lotus species are presented in Table 1 based on field observations and previous taxonomic studies, along with their general distribution and conservation status. In addition, we provided taxonomic keys and photo illustrations of snow lotus species, including habitats and closeups of the florets, pappus, phyllaries, and leaves (Fig. 1). Snow lotus species can be clearly distinguished based on their morphological characteristics. Moreover, we addressed the taxonomic notes of S. dorogostaiskii and S. krassnoborovii which are previously unresolved species. Based on the field surveys conducted in this study, in the early stages of flowering, the synflorescence was surrounded by pale green leaves, resembling cabbage. The synflorescence rachis became elongated and racemiform in mid-July after opening, and the uppermost leaves turned yellow, membranous, and boat-shaped. After late July, all the uppermost leaves withered. After pressing the specimens, membranous bracts were removed.

Morphological characteristics and geographic distribution map of snow lotus species in Central Asia and Southern Siberia. A General habitat. B Adult flower. C Inflorescences. D Disk flower. E Phyllaries. F Pappus. G Leaves. (Photo credit: S. Baasanmunkh and H.J. Choi)
Key to the snow lotus species
-
1.Capitula 5–30, strongly condensed corymbiform, sessile or with up to 3-cm-long peduncles; uppermost stem leaves stellate spreading
-
+Capitula 10–20, in racemiform, elongated synflorescence
-
2.Stem leaves lanceolate, apex long acuminate, with very dense glandular hair. Petiole remains of basal leaves with yellowish-brown strips up to 1 cm wide. Pappus straw-colored; phyllary triangular-ovate, apex acute or obtuse
-
S. orgaadayi
-
+Stem leaves narrowly ovate, elliptical, or obovate. Petiole remains of basal leaves with dark brown strips up to 2–3 mm wide. Bracts ovate elliptic, apex acute. Pappus dirty white colored
-
3.Stem leaves apex acute. Phyllaries triangular-ovate, glabrous, rarely sparsely pubescent apically or along the midvein
-
S. involucrata
-
+Stem leaves elliptical, apex obtuse. Phyllaries subulate to acuminate, densely pubescent middle-upper part
-
S. bogedaensis
-
4.Leaves oblong-elliptical to obovate, densely covered with glandular hair; upper stem leaves membranous, pale yellow, boat-shaped with strongly keeled, stellate surrounding synflorescence before flowering
-
S. dorogostaiskii
-
+Leaf blade narrowly ovate to elliptical, apex acute to acuminate, both surfaces light green, abaxially glabrous, adaxially sparsely villous to subglabrous. Synflorescence not enclosed by the uppermost leaves. Pappus straw colored
-
S. baicalensis
Assembly of plastome and ITS sequences
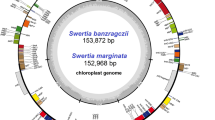
The plastomes of S. bogedaensis and S. dorogostaiskii were sequenced from Mongolia for the first time, and S. involucrata, S. orgaadayi, and S. baicalensis were resequenced from a new origin in Kyrgyzstan and Mongolia. De novo assembly generated a single contig for each sample (Table S2). The snow lotus plastomes exhibited a double-stranded circular DNA molecule 152,512–152,624 bp long (Table 2). It had a natural quadripartite structure that comprised a large single copy (LSC), a pair of inverted repeats (IRs), and a small single copy (SSC) (Fig. 2). The GC content in the plastomes was 37.7%, whereas the GC contents in the IR, LSC, and SSC regions were 43.1, 35.8, and 31.4–31.3%, respectively (Table 2). The snow lotus plastomes encoded 109 unique genes, including 4 rRNA, 25 tRNA, and 80 protein-coding (CDS) genes; among these, 4 rRNA genes, 7 tRNA genes, and 7 CDSs were duplicated in the IR regions (Table S3). They harbored 18 intron-containing genes, among which 16 had a single intron and 2 had two introns (Table S4). Four intron-containing genes were found to be duplicated in the IR region. The rps12 gene was a trans-spliced gene with the 5′ end located in the LSC and the 3′ end located in the IR.

Circular map of the complete chloroplast genome of A Saussurea dorogostaiskii and B Saussurea bogedaensis. The inner ring is divided into four areas, clockwise, and includes the SSC, IRb, LSC, and IRa. The genes in the outer ring region are transcribed clockwise, while those in the inner ring are transcribed counterclockwise. In addition, this figure also reflects the GC content; the inner ring in dark gray or green indicates the GC content, and the light gray or green indicates the AT content. In the lower left is a legend that classifies cp genes according to their functions
We newly sequenced 7 ITS sequences of snow lotus species, ranging from 650 to 700 bp in length. The final annotated plastome and ITS sequences were deposited in GenBank (Table 2 and S1).
Comparative analysis of the snow lotus plastome
In snow lotus plastomes, the genome structure, gene order, and boundaries between IRs and single-copy regions were similar. Co-linearity in gene placement among the five newly sequenced snow lotus plastomes was assessed using MAUVE (Fig. S1). Gene order and locations were conserved; however, the LSC region had a 20-kb inversion (between trnS-GCU and trnG-UCC). Additionally, another 3-kb translocation (between trnS-GCU and trnE-UUC) was detected within the snow lotus plastome. The sequence identities of the snow lotus were analyzed using mVISTA, with the S. obvallata (MH926128) plastome serving as a reference (Fig. S2). As expected, genic regions were more conserved than intergenic regions. This pair of IR regions was highly conserved, followed by the LSC and SSC regions. In snow lotus, IR lengths ranged from 25,193 to 25,220 bp, and the borders between the IR regions and the two single copy regions (LSC and SSC) were similar. The rps19 and ycf1 genes spanned the LSC/IRb and SSC/IRa junctions, respectively (Fig. S3).
The RSCU was computed for the 80 protein-coding genes of the snow lotus species. These CDSs contained 22,793–22,814 codons and encoded 20 amino acids in the plastome (Fig. S4A). Among them, leucine (10.53%) was the most frequently detected amino acid, whereas cysteine (1.09%) was the least ubiquitous (Fig. S4B). To identify codon patterns, we analyzed the codon distribution in eight plastomes. Snow lotus species exhibited similar patterns in 23 codons, and other codons have two types of patterns, with high RSCU values for AGA (arginine), TTA (leucine), and GCT (alanine) (Fig. S5). The first pattern is represented in S. bogedaensis, S. involucrate, and S. orgaadayi; other pattern is represented in S. dorogostaiskii and S. baicalensis. Almost all CDSs in snow lotus species have the standard ATG start codon, but psbL started with TGC/ACG. Codons with A or T in the third position had a strong codon bias. Among the three stop codons, TGA was the most commonly detected.
Distribution of repeat sequences
We investigated the repeat sequences of five Saussurea species to determine the characteristics and proportions of repeat sequences within the snow lotus group. The snow lotus species had a variable number of repeats, including forward, reverse, and palindromic repeats. For SSRs, mononucleotide motifs were the most abundant in all species, followed by tetranucleotide motif repeats (Fig. 3A). A total of 44–49 SSRs were identified, mostly in the LSC region, particularly within the intergenic spacer (IGS) region. Mononucleotide SSRs (25–31) mostly contained A (9–14), T (16–19), and C (1) repeat units. We found 26–46 tandem repeats that were generally 9–27 bp long (Fig. 3B). Among the protein-coding genes, rbcL and ycf2 commonly contain tandem repeats in the snow lotus plastomes. The 5′ end of the ndhB gene of S. baicalensis and S. dorogostaiskii included a 24-bp repeated insertion. A higher number of SSRs and tandem repeat sequences were observed in S. orgaadayi and S. dorogostaiskii, respectively (Fig. 3C). The snow lotus plastome included a higher number of forward repeats, whereas it had the lowest number of reverse repeats and no complementary repeats (Fig. 3D).

The distribution of repeat sequences in of snow lotus species and S. baicalensis plastomes. A The number of simple sequence repeat (SSR) motifs and the distribution of SSRs in the genome regions. B The number of repeat sequences, distribution of tandem repeats in the genome regions, and length of motifs. C Total number of repeat sequences of plastome. D Total length (bp) of repeat sequences of plastome
Estimation of genetic variability
We analyzed the genetic divergence of genes and intergenic regions within the snow lotus plastomes. Overall, the snow lotus plastomes had an average genetic diversity (Pi) value of 0.0016. The highest Pi values were observed in IGS regions ndhJ–ndhK (0.018) and ndhD-psaC (0.016) in the LSC and SSC regions, respectively. Genic and intergenic regions in the IR regions were relatively constant (Fig. 4).

Comparison of the nucleotide diversity (Pi) values among the snow lotus species. The mean Pi value indicated by blue line
Molecular evolution of snow lotus
A total of 80 consensus protein-coding genes from eight snow lotus samples were evaluated to assess selective pressure, and compared with S. obvallata. Three genes were found to have undergone positive selection (Fig. 5): ycf2 had undergone positive selection in most snow lotus species, except S. bogedaensis MGL and S. involucrata MGL; ndhB (1.83) was found in S. baicalensis MGL and S. dorogostaiskii MGL; rpoA (1.95) was found in S. orgaadayi. Fifteen genes were found to be under purifying selection in snow lotus species. Some of them show specific selection in a single species: psbT in S. involucrata KGZ, rps2 and ycf4 in S. baicalensis CHN, and ccsA in S. bogedaensis MGL. Furthermore, synonymous substitution (Ks) was represented in 21 genes, with the highest values in ndhB and rpoC2 in the S. baicalensis and S. dorogostaiskii, and ndhF and psbB in S. involucrata from Kyrgyzstan (Table S5). The psaA, psbC, ndhB, ndhI, and rpoC2 had specific mutations in S. dorogostaiskii and S. baicalensis plastomes. Moreover, psaB, psbB, and ndhD included specific mutations in the S. bogeadensis, S. involucrata, and S. orgaadayi plastomes. In the 43 genes that included non-synonymous substitutions (Ka), high values were noted in ndhB and rpoC2 of S. baicalensis and S. dorogostaiskii, and psbB of S. involucrata from Kyrgyzstan (Table S6). Among the noticed genes, six (ndhJ, psaA, psbA, psbH, rps4, and ycf2) exhibited the same substitution in all species; eight (accD, atpB, matK, psaC, rbcL, rpoC1, rpoC2, and ycf1) were highly mutated in S. baicalensis and S. dorogostaiskii; nine (ndhA, ndhB, psaJ, rps15, rps16, rpl20, rpl33, and ycf4) included mutation in only S. baicalensis and S. dorogostaiskii; six genes (psaB, psbB, psbL, psbT, ndhD, rpl22) include mutations in only S. bogeadensis, S. involucrata and S. orgaadayi; and other genes included species-specific mutations.

Positively selected genes (Ka/Ks > 1) in the snow lotus species and S. baicalensis
Phylogenic position of snow lotus group within Saussurea
Two datasets (plastome and nrDNA) were compiled to verify the phylogenetic position of the snow lotus species. The datasets contained 19 plastomes and 32 ITS sequences of Saussurea species from four different subgenus according to Raab-Straube (2003), Kita et al. (2004), Wang et al. (2007; 2010), Chen and Wang (2018), Chen et al. (2019), and Baasanmunkh et al. (2020), including those sequenced samples in this study (Table S7). Jurinea multiflora (L.) B.Fedtsch. and Arctium lappa L. were used as outgroups. The plastome alignment was 156,494 bp long, of which 154,497 bp were constant and 633 bp were parsimony-informative. The ITS alignment contained 442 bp, including 283 and 76 bp with constant and parsimony-informative characteristics, respectively. The newly sequenced S. dorogostaiskii samples from both Mongolia and Russia (previously named S. krasnoborovii) formed a highly supported monophyletic clade (BP = 100%); S. orgaadayi samples from Khangai (previously named S. involucrata) and Mongolian Altai in Mongolia were identified as the same species as S. orgaadayi from Xinjiang, China; and S. bogedaensis samples from Gobi-Altai (previously named S. involucrata) and Dzungarian Gobi regions in Mongolia were identified as the same species as S. bogedaensis from Xinjiang, China (Fig. 6). The topologies of the BI, ML, and MP analyses were highly congruent with the same dataset and supported by strong bootstrap values and PPs. The phylogenetic positions of the sections within Saussurea were not consistent among the phylogenetic tree of plastomes and nrDNAs. S. bogedaensis, S. involucrata, and S. orgaadayi were clustered with section Amphilaena species; S. dorogostaiskii and S. baicalensis clustered together under subgenus Saussurea based on the plastome sequences (Fig. 6A). Whereas in the ITS phylogenetic tree, the snow lotus species exhibited a monophyletic pattern (Fig. 6B).

Phylogenetic tree based on A plastome and B nrDNA. Bayesian inference (BI), maximum likelihood (ML) and maximum parsimony (MP) support values indicated at each branch, following the order: BI/ML/MP. Maximum support values are labeled by a star. Black colored numbers indicate nodes constrained with fossils in the analysis. The newly sequenced samples are in bold. The samples from China, Kyrgyzstan, and Mongolia are abbreviated as CHN, KZS, and MNG, respectively. The branch of subg. Ericoryne, subg. Saussurea, subg. Amphilaena, and subg. Theodorea are represented by orange, green, blue, and purple color, respectively
Discussion
Classification and distribution of the snow lotus species
Taxonomic identification of Saussurea is notoriously difficult because of the high diversity of specialized morphology traits that have developed to adapt them to the different environmental stresses experienced in mountainous regions (Wang et al. 2009; Zhang et al. 2019; Zhang et al. 2021a, c). For example, Amphilaena is a non-monophyletic group based on DNA barcoding and plastome data (Wang et al. 2009; Xu et al. 2019). Recent whole-genome sequencing techniques and the sampling of threatened species have increased the availability of DNA barcoding (Li et al. 2019). In this study, we investigated the classification of snow lotus species by comparing our findings with those of previous studies. In particular, some specimens that were previously identified as S. dorogostaiskii (Smirnov 2004) were identified as S. krasnoborovii in this study; hence, we preserved the name S. dorogostaiskii for populations from Mongolia without membranous, pale-yellow upper stem leaves. More recently, S. krasnoborovii has been classified as the same as S. dorogostaiskii based on morphological characteristics (Raab-Straube 2017). Later, Smirnov et al. (2018) updated the checklist of Saussurea in Eurasia, which classified S. dorogostaiskii and S. krasnoborovii as different species. During our field surveys, we found various habitat types of S. dorogostaiskii in Mongolia and Russia at different periods, all of which were identified as single species based on their morphology, plastome, and nrDNA sequences (Table 1 and Figs. 1 and 6). Our observations confirmed the results of Raab-Straube (2017) that S. krasnoborovii is synonym of S. dorogostaiskii. This species is distributed in the Buryat and Tyva of Russia, and the Khuvsgul region of Mongolia (Fig. 1K). This species grows on both open and forested mountains between 1500 and 2500 m, primarily in and around boulders or talus patches, and sometimes within smaller scree rocks (Norris 2020).
Saussurea baicalensis is a type species of section Pycnocephala subgenus Saussurea (Shi and Raab-Straube 2011), which is closely related to S. dorogostaiskii (Fig. 6). Initially, S. dorogostaiskii shared a morphology similar to S. baicalensis (Palibin 1928) and was classified in the same section as S. baicalensis based on morphology (Smirnov 2004). Raab-Straube (2017) later discovered that S. dorogostaiskii occupied an intermediate position between the section Amphilaena subgenus Amphilaena and section Pycnocephala (Lagurostemon) subgenus Saussurea (Table 3). Consequently, S. dorogostaiskii belongs to section. Pycnocephala with S. baicalensis based on their genetic and morphological features. Further studies focusing on determining the position of section Pycnocephala within this genus are required, because the phylogenetic position of section Pycnocephala on the plastome and nrDNA trees is not consistent. The section Pycnocephala was clustered with subgenus Saussurea species on the plastome tree (Fig. 6A), and with subgenus Amphilaena species in the nrDNA tree (Fig. 6B). This section may have an intermediate position between the subgenuses Amphilaena and Saussurea.
The morphological features of S. orgaadayi, S. involucrata, and S. bogedaensis are very similar, such as bracts surrounding the corymbiform synflorescence, cream-yellow bracts that aggregate below the florescence, and hollow stem ≥ 1.5 cm in diameter near the base (Fig. 1) (Shi and Raab-Straube 2011; Raab-Straube 2017; Chen and Wang 2018). However, they are significantly differentiated by the shape of the bract, pappus, and leaf margin (Table 1) (Chen and Wang 2018). Based on our molecular and morphological analysis, we confirmed the distribution of S. bogedaensis in the Gobi-Altai region and S. orgaadayi in the Khangai region of Mongolia, where S. involucrata was previously observed (Grubov 1982). The samples from the Khangai and Altai Mountains present straw-colored pappus, triangular-ovate bracts with a long acuminate apex, and phyllaries densely pubescent throughout and with up to 60-cm-tall stems, which are typical characteristics of S. orgaadayi rather than S. involucrata (Shi and Raab-Straube 2011). The samples from Gobi-Altai presented elliptic stem leaves with an obtuse apex and dirty white-colored pappus, which are typical characteristics of S. bogedaensis (Chen and Wang 2018). As a result, S. involucrata was excluded from the Mongolian flora because of long-term misidentification as S. orgaadayi or S. bogedaensis. In addition, recently published studies have suggested that S. involucrata were distributed in the western Tianshan Mountains in China and Kyrgyzstan, S. bogedaensis in eastern Tiansan in China, Dzungarian Gobi and Govi-Altai regions in Mongolia, and S. orgaadayi in the Altai Mountains in China, Mongolia, and Russia (Fig. 1K) (Raab-Straube 2017; Chen and Wang 2018; Chen et al. 2019; Baasanmunkh et al. 2020; Erst et al. 2022).
Plastome feature and diversity
The complete plastome of Saussurea has recently been studied (Xu et al. 2019; Zhang et al. 2019, 2021a, c). However, previous studies have not explored snow lotus species in Central Asia and Southern Siberia, and there remains a gap for a better understanding of their relationships and evolutionary patterns. In this study, we sequenced and characterized the plastomes of snow lotus species in Central Asia and Southern Siberia. The complete plastome of the snow lotus is a typical quadripartite structure with one LSC, one SSC, and two IR regions (Fig. 2), which is a highly conserved pattern in green plants (Bruneau et al. 2007). Plastome genome size and gene order and number are conserved in the snow lotus plastome, including 4 rRNA, 25 tRNA, and 80 protein-coding genes. The snow lotus plastome included a 20-kb inversion and 3-kb translocation in the LSC region (Fig. S1). Such inversions have been observed in most Asteraceae (Zhang et al. 2019). No additional specific inversions were observed among the studied species. The IRb/SSC and IRa/LSC boundary shifts between the species commonly included the ycf1 and rps19 pseudogenes, respectively. Overall, our results demonstrated that the conservation pattern of plastomes is similar to those of other Saussurea species (Zhang et al. 2019; Yun and Kim 2022).
Many studies have reported the use of chloroplast SSR markers with high polymorphisms in Saussurea (Shen et al. 2020; Yun and Kim 2022). In previous studies, the genetic variability in snow lotus species has been investigated using genetic markers of the nuclear genome (Yuan et al. 2009; Wei et al. 2017; Nyamgerel et al. 2023), but they revealed lower genetic diversity. In this study, we found 49 SSRs (Fig. 3A), among these, 38 chloroplast SSRs ≥ 10 bp that can be used in population genetic studies (Table S8). These markers can aid in a better understanding of the genetic diversity and distribution patterns of snow lotus populations.
Previous phylogenetic studies of snow lotus species have mainly used nrDNA (ITS) and plastid (matK, rbcL, trnK, trnH-psbA) barcoding markers (Chen and Wang 2018; Chen et al. 2019; Baasanmunkh et al. 2020). However, based on the plastome analysis of Saussurea, these plastid markers failed to resolve relationships across the genus (Zhang et al. 2019). Our study also revealed a relatively low nucleotide diversity in these four regions (Fig. 4). We identified two relatively variable regions (ndhJ-ndhK and ndhD-psaC). Further barcoding studies of these two intergenic regions, combined with nuclear ITS markers, are needed to identify snow lotus species.
Molecule evolution of the snow lotus group
Global cooling since the middle of the Miocene has affected Saussurea species in a wide variety of habitats, including cold and dry alpine meadows, steppe deserts, and screes (Xu et al. 2019; Zhang et al. 2021a). For example, the snow lotus species includes one of the most diverse groups within Saussurea that grow at high altitudes (Chen et al. 2019; Baasanmunkh et al. 2020). Understanding how different species adapt to extreme environments is an extension of the main goal of evolutionary biology (Zhang et al. 2021b). In this study, we estimated the selection pressure (Ka/Ks) of protein-coding genes in the snow lotus species, which has long been considered an indicator of adaptive evolution in green plants (Kimura 1979). Genes with a Ka/Ks > 1 ratio were considered under positive selection, and genes with a Ka/Ks ratio < 1 were considered under purifying selection (Yang and Bielawski 2000). Most protein-coding genes in Saussurea are under purifying selection (Zhang et al. 2019; Yun and Kim 2022). Among the snow lotus species, three genes were found to be under positive selection (Fig. 5). These genes may play important roles in the adaptation of the snow lotus to high altitudes. Among these, ycf2 is open reading frame in most higher plants that encodes proteins necessary for cell survival (Drescher et al. 2000). The rpoA gene encodes the α-subunit of RNA polymerase type I (plastid-encoded polymerase (PEP)) (Little and Hallick 1988). The rpo genes are relatively fast-evolving sequences that have been used as markers in phylogenetic studies (Krawczyk and Sawicki 2013). In addition, a photosynthesis-related gene, ndhB, was discovered in S. baicalensis and S. dorogostaiskii, which is associated with photosystem I to form a super-complex that mediates cyclic electron transport (Munekage et al. 2004).
Interestingly, the selective pressure on protein-coding genes in S. dorogostaiskii is similar to that in S. baicalensis, compared to other snow lotus species (Fig. 5). This result corresponds with the morphological features of S. dorogostaiskii. It is distinguished from the others by its elongated racemiform synflorescence and uppermost leaves, which are located on branches up to 1 m high (Table 2 and Fig. 1). S. baicalensis also exhibits racemiform synflorescences (Shi and Raab-Straube 2011). Other snow lotus species exhibit corymbiform synflorescences, which may be derived from racemiform synflorescences (Raab-Straube 2017). Furthermore, non-synonymous mutations of protein-coding genes were demonstrated in three types of patterns among the snow lotus species (Table S6): the first group was represented in S. dorogostaiskii and S. baicalensis, the second group was represented in S. bogedaensis and S. involucrata, and the third group included S. orgaadayi from China and Mongolia (Altai and Khangai Mountains). Within the second group, S. involucrata samples from China and Kyrgyzstan had various nucleotide substitutes, and S. bogedaensis (Mongolia) and S. involucrata (China) were relatively similar than S. involucrata (Kyrgyzstan). Further studies of the adaptation of the snow lotus species are required. In addition, S. involucrata (China) and S. bogedaensis (Mongolia) are most recently separated from S. involucrata (Kyrgyzstan) on the plastome analysis. They may share a common adaptation because of the similar conditions of environment as S. involucrata (Kyrgyzstan).
Conclusion
The present study ascertained the nomenclature, phylogeny, and plastome evolution of the snow lotus species in China, Kyrgyzstan, Mongolia, and Russia. In particular, S. involucrata was excluded from the Mongolian flora, and S. krasnoborovii was synonymous with S. dorogostaiskii. The geographic distribution of each snow lotus was separated into the Altai, Tienshian, and Sayan Mountains. Snow lotus has a conserved plastome structure and gene content similar to those of most Saussurea. However, they have few genetic divergence regions and SSR markers that are suitable for identifying population genetic structure and gene flow patterns. In addition, S. dorogostaiskii is closely related to S. baicalensis, compared with other snow lotus species, and they have similar molecular adaptation patterns. This may have been caused by nonrandom recombination associated with climate change, making it an interesting topic for future evolutionary investigation.
Data availability
The datasets generated and/or analyzed in this study are available in GenBank, National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov/genbank/) under the accession numbers of plastome: OR426625-OR426629 and ITS: OR673962, ON394565, ON399085, ON399084, ON399088, OQ826667, and OQ826670.
References
Amiryousefi A, Hyvönen J, Poczai P (2018) IRscope: an online program to visualize the junction sites of chloroplast genomes. Bioinformatics 34(17):3030–3031. https://doi.org/10.1093/bioinformatics/bty220
Antil S, Abraham JS, Sripoorna S et al (2023) DNA barcoding, an effective tool for species identification: a review. Mol Biol Rep 50(1):761–775. https://doi.org/10.1007/s11033-022-08015-7
Baasanmunkh S, Nyamgerel N, Bayarmaa G-A et al (2020) A new record of critically endangered Saussurea bogedaensis (Asteraceae) from Dzungarian Gobi, Mongolia. PhytoKeys 160:109–121. https://doi.org/10.3897/phytokeys.160.55603
Baasanmunkh S, Urgamal M, Oyuntsetseg B et al (2022) Flora of Mongolia: annotated checklist of native vascular plants. PhytoKeys 192:63–169. https://doi.org/10.3897/phytokeys.192.79702
Babicki S, Arndt D, Marcu A et al (2016) Heatmapper: web-enabled heat mapping for all. Nucleic Acids Res 44:W147–W153. https://doi.org/10.1093/nar/gkw419
Beier S, Thiel T, Münch T et al (2017) MISA-web: a web server for microsatellite prediction. Bioinformatics 33(16):2583–2585. https://doi.org/10.1093/bioinformatics/btx198
Benson G (1999) Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27(2):573–580. https://doi.org/10.1093/nar/27.2.573
Bock DG, Andrew RL, Rieseberg LH (2014) On the adaptive value of cytoplasmic genomes in plants. Mol Ecol 23(20):4899–4911. https://doi.org/10.1111/mec.12920
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30(15):2114–2120. https://doi.org/10.1093/bioinformatics/btu170
Brudno M, Malde S, Poliakov A et al (2003) Glocal alignment: finding rearrangements during alignment. Bioinformatics 19(1):54–62. https://doi.org/10.1093/bioinformatics/btg1005
Bruneau A, Starr JR, Joly S (2007) Phylogenetic relationships in the genus Rosa: new evidence from chloroplast DNA sequences and an appraisal of current knowledge. Syst Bot 32(2):366–378. https://doi.org/10.1600/036364407781179653
Chen YS (2015) Asteraceae II (Saussurea): part 2: flora of the Pan-Himalaya. Cambridge Universiry Press, Cambridge
Chen J, Wang YJ (2018) New Saussurea (Asteraceae) species from Bogeda Mountain, Eastern Tianshan, China, and inference of its evolutionary history and medical usage. PLoS ONE 13(7):e0199416. https://doi.org/10.1371/journal.pone.0199416
Chen J, Zhao YB, Wang YJ, Li XG (2019) Identification of species and materia medica within Saussurea subg Amphilaena Based on DNA Barcodes. Peerj 7:e6357. https://doi.org/10.7717/peerj.6357
Chik WI, Zhu L, Fan LL et al (2015) Saussurea involucrata: a review of the botany, phytochemistry and ethnopharmacology of a rare traditional herbal medicine. J Ethnopharmacol 172:44–60. https://doi.org/10.1016/j.jep.2015.06.033
Darling AE, Mau B, Perna NT (2010) progressiveMauve: Multiple Genome Alignment with gene gain, loss and rearrangement. PLoS ONE 5(6):e11147. https://doi.org/10.1371/journal.pone.0011147
Darriba D, Taboada GL, Doallo R, Posada D (2012) jModelTest 2: more models, new heuristics and parallel computing. Nat Methods 9(8):772–772. https://doi.org/10.1038/nmeth.2109
Doyle JJ, Doyle JL (1987) A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull 19:11–15
Drescher A, Ruf S, Calsa T Jr et al (2000) The two largest chloroplast genome-encoded open reading frames of higher plants are essential genes. Plant J 22(2):97–104. https://doi.org/10.1046/j.1365-313x.2000.00722.x
Erst AS, Nikulin AYU, Nikulin VYU et al (2022) Distribution analysis, updated checklist, and DNA barcodes of the endemic vascular flora of the Altai mountains, a Siberian biodiversity hotspot. Syst Biodivers 20(1):1–30. https://doi.org/10.1080/14772000.2022.2049391
ESRI (2011) ArcGIS desktop: release 10.2. Environmental Systems Research Institute: Redlands, CA, USA
Frazer KA, Pachter L, Poliakov A et al (2004) VISTA: computational tools for comparative genomics. Nucleic Acids Res 32(2):273–279. https://doi.org/10.1093/nar/gkh458
Grubov VI (1982) Key to the vascular plants of Mongolia (with an atlas). Nauka, Leningrad
Hall T, Biosciences I, Carlsbad C (2011) BioEdit: an important software for molecular biology. GERF Bull Biosci 2(1):60–61
Jansen RK, Cai Z, Raubeson LA et al (2007) Analysis of 81 genes from 64 plastid genomes resolves relationships in Angiosperms and identifies genome-scale evolutionary patterns. Proc Natl Acad Sci U S A 104(49):19369–19374. https://doi.org/10.1073/pnas.0709121104
Katoh K (2002) MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30(14):3059–3066. https://doi.org/10.1093/nar/gkf436
Kimura M (1979) Model of effectively neutral mutations in which selective constraint is incorporated. Proc Natl Acad Sci U S A 76(7):3440–3444. https://doi.org/10.1073/pnas.76.7.3440
Kita Y, Fujikawa K, Ito M et al (2004) Molecular phylogenetic analyses and systematics of the genus Saussurea and related genera (Asteraceae, Cardueae). Taxon 53:679–690. https://doi.org/10.2307/4135443
Krawczyk K, Sawicki J (2013) The uneven rate of the molecular evolution of gene sequences of DNA-dependent RNA polymerase I of the genus Lamium L. Int J Mol Sci 14(6):11376–11391. https://doi.org/10.3390/ijms140611376
Kurtz S (2001) REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res 29(22):4633–4642. https://doi.org/10.1093/nar/29.22.4633
Lagesen K, Hallin P, Rødland EA et al (2007) RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res 35(9):3100–3108
Li HT, Yi TS, Gao LM et al (2019) Origin of angiosperms and the puzzle of the Jurassic gap. Nat Plants 5(5):461–470. https://doi.org/10.1038/s41477-019-0421-0
Lipschitz S (1979) Genus Saussurea DC. (Asteraceae). Nauka, Leningrad
Little MC, Hallick RB (1988) Chloroplast rpoA, rpoB, and rpoC genes specify at least three components of a chloroplast DNA-dependent RNA polymerase active in tRNA and mRNA transcription. J Biol Chem 263(28):14302–14307. https://doi.org/10.1016/s0021-9258(18)68221-3
Long EO, Dawid IB (1980) Repeated genes in Eukaryotes. Annu Rev Biochem 49:727–764
Lowe TM, Chan PP (2016) tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res 44(1):W54–W57
Munekage Y, Hashimoto M, Miyake C et al (2004) Cyclic electron flow around photosystem I is essential for photosynthesis. Nature 429(6991):579–582
Nicolas D, Patrick M, Guillaume S (2017) NOVOPlasty: de novo assembly of organelle 558 genomes from whole genome data. Nucleic Acids Res 45:e18. https://doi.org/10.1093/nar/gkw955
Norris J (2020) Synthesis of the local knowledge and ecology of vansemberuu (Saussurea dorogostaiskii Palib.) for conservation, Darkhad Valley, Khuvsgul. University of Vermont Honors College, Mongolia
Nyambayar D, Oyuntsetseg B, Tungalaga R (2011) Mongolian red list and conservation action plans of plants, Ulaanbaatar
Nyamgerel N, Baasanmunkh S, Oyuntsetseg B et al (2023) Genetic diversity of the threatened Saussurea dorogostaiskii (Asteraceae) in the Khuvsgul region of Mongolia. Korean J Plant Taxon 53(1):14–24. https://doi.org/10.11110/kjpt.2023.53.1.14
Omori Y, Takayama H, Ohba H (2000) Selective light transmittance of translucent bracts in the Himalayan giant glasshouse plant Rheum nobile Hook.f. & Thomson (Polygonaceae). Bot J Linn Soc 132(1):19–27. https://doi.org/10.1111/j.1095-8339.2000.tb01852.x
Palibin IV (1928) New Saussurea from northern Mongolia. Russ Bot J 13:106–111 [in Russian]
Raab-Straube E (2003) Phylogenetic relationships in Saussurea (Compositae, Cardueae) sensu lato, inferred from morphological, ITS and trnL-trnF sequence data, with a synopsis of Himalaiella gen. nov., Lipschitziella and Frolovia. Willdenowia 33:379–402. https://doi.org/10.3372/wi.33.33214
Raab-Straube E (2017) Taxonomic revision of Saussurea subgenus Amphilaena (Compositae, Cardueae). BGBM, Berlin
Rambaut A (2012) FigTree v1.4. Molecular evolution, phylogenetics and epidemiology. University of Edinburgh, Institute of Evolutionary Biology. https://tree.bio.ed.ac.uk/software/figtree
Ravi V, Khurana JP, Tyagi AK, Khurana P (2008) An update on chloroplast genomes. Plant Syst Evol 271:101–122. https://doi.org/10.1007/s00606-007-0608-0
Rodnina MV, Beringer M, Wintermeyer W (2007) How ribosomes make peptide bonds. Trends Biochem Sci 32(1):20–26. https://doi.org/10.1016/j.tibs.2006.11.007
Ronquist F, Teslenko M, van der Mark P et al (2012) MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol 61(3):539–542. https://doi.org/10.1093/sysbio/sys029
Rozas J, Ferrer-Mata A, Sánchez-DelBarrio JC et al (2017) DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol Biol Evol 34(12):3299–3302. https://doi.org/10.1093/molbev/msx248
Shen J, Zhang X, Landis JB et al (2020) Plastome evolution in Dolomiaea (Asteraceae, Cardueae) using phylogenomic and comparative analyses. Front Plant Sci 11:1–15. https://doi.org/10.3389/fpls.2020.00376
Shi Z, Raab-Straube E (2011) Saussurea Candolle. In: Wu ZY, Raven PH (eds) Flora of China. Science Press, Beijing pp 56–149
Smirnov SV (2004) Notes on the genus Saussurea DC. (Asteraceae) in Altai. Turczaninowia 7:11–17
Smirnov SV, Kechaykin AA, Sinitsyna TA et al (2018) Synopsis of the genus Saussurea DC. (Asteraceae) of Euroasia. Ukranian J Ecol 8:270–285
Stamatakis A (2014) RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30(9):1312–1313. https://doi.org/10.1093/bioinformatics/btu033
Swofford DL, Documentation B (1989) Phylogenetic analysis using parsimony Illinois Natural History Survey: Champaign, IL, USA
Talavera G, Castresana J (2007) Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol 56(4):564–577. https://doi.org/10.1080/10635150701472164
Tamura K, Stecher G, Kumar S (2021) MEGA11: molecular evolutionary genetics analysis version 11. Mol Biol Evol 38:3022–3027
Thompson JD, Gibson TJ, Higgins DG (2003) Multiple sequence alignment using ClustalW and ClustalX. Curr Protoc Bioinforma 1:2–3. https://doi.org/10.1002/0471250953.bi0203s00
Tillich M, Lehwark P, Pellizzer T et al (2017) GeSeq—versatile and accurate annotation of organelle genomes. Nucleic Acids Res 45(1):W6–W11. https://doi.org/10.1093/nar/gkx391
Wang YJ, Liu JQ, Miehe G (2007) Phylogenetic origins of the Himalayan endemic Dolomiaea, Diplazoptilon and Xanthopappus (Asteraceae: Cardueae) based on three DNA regions. Ann Bot 99:311–322. https://doi.org/10.1093/aob/mcl259
Wang YJ, Susanna A, Von Raab-Straube E et al (2009) Island-like radiation of Saussurea (Asteraceae: Cardueae) triggered by uplifts of the Qinghai-Tibetan Plateau. Biol J Linn Soc 97(4):893–903. https://doi.org/10.1111/j.1095-8312.2009.01225.x
Wang D, Zhang Y, Zhang Z et al (2010) KaKs_Calculator 2.0: a toolkit incorporating gamma-series methods and sliding window strategies. GPB 8(1):77–80. https://doi.org/10.1016/S1672-0229(10)60008-3
Wei S, Yang W, Wang X, Hou Y (2017) High genetic diversity in an endangered medicinal plant, Saussurea involucrata (Saussurea, Asteraceae), in western Tianshan Mountains, China. Conserv Genet 18:1435–1447. https://doi.org/10.1007/s10592-017-0991-1
White TJ, Bruns T, Lee S, Taylor J (1990) Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. PCR Protoc 18(1):315–322. https://doi.org/10.1016/b978-0-12-372180-8.50042-1
Xu LS, Herrando-Moraira S, Susanna A et al (2019) Phylogeny, origin and dispersal of Saussurea (Asteraceae) based on chloroplast genome data. Mol Phylogenet Evol 141:106613. https://doi.org/10.1016/j.ympev.2019.106613
Yang Z, Bielawski JR (2000) Statistical methods for detecting molecular adaptation. Trends Ecol Evol 15(12):496–503. https://doi.org/10.1016/S0169-5347(00)01994-7
Yuan XF, Dai ZH, Wang XD, Zhao B (2009) Assessment of genetic stability in tissue-cultured products and seedlings of Saussurea involucrata by RAPD and ISSR markers. Biotechnol Lett 31:1279–1287. https://doi.org/10.1007/s10529-009-9984-6
Yun S, Kim SC (2022) Comparative plastomes and phylogenetic analysis of seven Korean endemic Saussurea (Asteraceae). BMC Plant Biol 22(1):550. https://doi.org/10.1186/s12870-022-03946-6
Zhang X, Deng T, Moore MJ et al (2019) Plastome phylogenomics of Saussurea (Asteraceae: Cardueae). BMC Plant Biol 19(1):1–10. https://doi.org/10.1186/s12870-019-1896-6
Zhang X, Landis JB, Sun Y et al (2021a) Macroevolutionary pattern of Saussurea (Asteraceae) provides insights into the drivers of radiating diversification. Proc R Soc B Biol Sci 288(1962):20211575. https://doi.org/10.1098/rspb.2021.1575
Zhang X, Sun Y, Landis JB et al (2021b) Transcriptomes of Saussurea (Asteraceae) provide insights into high-altitude adaptation. Plants 10(8):1–14. https://doi.org/10.3390/plants10081715
Zhang Y, Chen J, Sun H (2021c) Alpine speciation and morphological innovations: revelations from a species-rich genus in the northern hemisphere. AoB Plants 13(3):1–12. https://doi.org/10.1093/aobpla/plab018
Zheng S, Poczai P, Hyvönen J et al (2020) Chloroplot: an online program for the versatile plotting of organelle genomes. Front Genet 11:576124. https://doi.org/10.3389/fgene.2020.576124
Acknowledgements
This study is a part of the first author’s PhD thesis.
Funding
This work was supported by the Korea National Arboretum (Grant No. KNA1-1–26, 20–1) and Korea Basic Science Institute (Nantional Research Facilities and Equipment Center) grant funded by the Ministry of Education (Grant No. 2023R1A6C101B022).
Author information
Authors and Affiliations
Contributions
NN, SB, and HJC conceived and designed the study; NN conducted the experiment; GB, ZT, and IP provided experimental support; SB, BO, GB, GL, EP, HYG, and HJC collected the samples; NN and SB prepared the original draft; IP and HJC revised and finalized the manuscript. All authors have read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nyamgerel, N., Baasanmunkh, S., Oyuntsetseg, B. et al. Comparative plastome analysis and taxonomic classification of snow lotus species (Saussurea, Asteraceae) in Central Asia and Southern Siberia. Funct Integr Genomics 24, 42 (2024). https://doi.org/10.1007/s10142-024-01309-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10142-024-01309-y