
Abstract
Among human CD8+ T cells, senescent cells are marked by the expression of CD57. The frequency of senescent CD57+CD8+ T cells is significantly correlated with aging and age-associated disorders, and it can be measured by multi-color flow cytometry. However, multi-color flow cytometry presents challenges in terms of accessibility and requires significant resource allocation. Therefore, developing a rapid and straightforward method for quantifying CD57+CD8+ T cells remains a key challenge. This study introduces a microfluidic device composed of a PDMS microfluidic channel with a pre-modified glass substrate for anti-CD8 antibody immobilization. This design allows blood samples to flow through, enabling the selective capture of CD8+ T cells while minimizing the required blood sample volume. This technology enables accurate and reliable quantification of CD57+ cells among captured CD8+ T cells through fluorescence image analysis. The ability of the device to easily quantify senescent CD57+CD8+ T cells is anticipated to contribute significantly to both immunological research and clinical applications.
Similar content being viewed by others

1 Introduction
Both adaptive and innate immunities are influenced by aging, but the most notable changes can be observed in the CD8+ T cell population [1]. During the late stage of differentiation, CD8+ T cells exhibit a lack of surface expression of co-stimulatory molecules such as CD27 or CD28. Moreover, acquiring CD57 expression is considered an irreversible step towards differentiation into ‘senescent’ cells. These cells have reduced proliferative capacity and altered functional properties [2, 3]. In patients chronically infected with cytomegalovirus or human immunodeficiency virus, the increased frequency of senescent CD57+CD8+ T cells that produce pro-inflammatory mediators is associated with the early onset of cardiovascular diseases [4]. Furthermore, substantial evidence suggests an accumulation of CD57+CD8+ T cells in patients with chronic inflammatory diseases linked with aging [5,6,7,8]. A longitudinal analysis has demonstrated marked age-dependent changes in the frequency of the CD57+CD8+ T-cell subset in peripheral blood [9].
Identifying senescent CD57+CD8+ T cells from blood samples relies on acquiring surface phenotype data by flow cytometry. However, the access to and availability of multi-color flow cytometers are often limited in most clinical situations. Small in-house laboratories may lack the resources to train and maintain staff with the necessary expertise and to provide specialized equipment and reagents used for flow cytometric assessments. Moreover, flow cytometric data can be influenced by the proficiency of the operator and the physical properties of the flow cytometer and fluorescent dyes [10]. These can pose a challenge for operating a flow cytometer, and there remains an unmet need to develop a rapid and straightforward method for quantifying CD57+CD8+ T cells.
Several microfluidic methods have been proposed to detect and analyze the protein expression on the cell surface [11,12,13,14,15]. One approach involves capturing the cells of interest using an antibody immobilized on the surface of the microfluidic channel. In contrast, non-target cells that only weakly interact with the channel surface are washed away. The captured cells can then be collected and subjected to further analysis by applying stronger shear forces, introducing air bubbles, or using specific enzymes to detach the cells from the device [16,17,18,19]. The effectiveness and purity of this cell capture device are influenced by various factors, such as flow rates, shear stress conditions, and the shape of the channel [20]. Micropost arrays are often used to enhance the capture efficiency by enlarging the capture area [21,22,23,24,25]. Another strategy is to use a herringbone mixer, which facilitates surface contact between the cells and capture molecules [26, 27]. However, complex channel structures, such as microposts and herringbone patterns, can negatively affect the microscopic measurement area, potentially interfering with the fluorescence imaging analysis.
In this study, we present a microfluidic capture device designed to quantify senescent CD57+CD8+ cells among CD8+ T cells directly from whole blood samples. The microfluidic channel was optimized to ensure the capture of cells expressing surface CD8 molecules without clogging from unwanted cells. This involved fine-tuning the flow rate and capture duration. Using a flat glass substrate provides a robust capability to capture hundreds of CD8+ T cells from either concentrated white blood cells (WBCs) or whole blood samples. This can be achieved by simply injecting a sample as small as tens of microliters. The captured cells were labeled with fluorescent antibodies, and the CD57+CD8+/CD8+ T-cell ratios were conveniently measured. We validated the device by comparing the frequency of CD57+CD8+ T cells with conventional flow cytometry results. Our microfluidic method facilitates accurate and reliable analysis of T cell surface marker expressions in a fast and simple manner without trained professionals. Notably, the blood sample volume required for this method is as small as 10 µL, which can lead to an on-site analysis platform using the blood samples collected by fingerstick. With its superior performance, our microfluidic method is expected to be widely used for both immunological research field and practical clinical applications.
2 Results and Discussion
2.1 Microfluidic CD8+ T-Cell Capture
2.1.1 Principles of Operation
A microfluidic device for affinity-based cell capture was developed to capture CD8+ T cells from blood samples and to analyze the surface CD57 expression (Fig. 1). The microfluidic device was constructed by assembling a simple Polydimethylsiloxane (PMDS) microfluidic channel and a glass substrate that was pre-modified for antibody immobilization. The glass substrate was coated with polyethylene glycol (PEG) and biotin-PEG [28,29,30,31] to enable target-specific antibodies to be easily immobilized after the microfluidic channel was assembled. Conventional oxygen plasma bonding of the PDMS and glass substrate can damage the biotin-PEG coating. We used vacuum-assisted assembly of the microfluidic channel to avoid damaging the PEG surface [32]. The PDMS channel and the glass substrate were assembled by applying negative pressure on the surrounding channels (vacuum line). The capture antibody for the CD8 molecule was immobilized by sequentially flowing neutravidin and biotinylated antibodies. Then, a blood sample was injected, and CD8+ T cells were captured. Finally, fluorescently labeled anti-CD8 and anti-CD57 antibodies were flowed over the channel. The detailed procedures are described with discussion in the following sections.

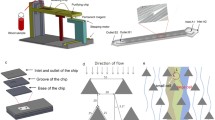
Microfluidic device preparation and T-cell capture/analysis procedure. a The PDMS microfluidic channel design and fabrication. b Preparation of the glass slide with the PEG and Biotin-PEG coating. c Antibody immobilization, CD8+ T-cell capture and fluorescent labeling procedure. d A microfluidic device and setup. e Microscopic images of cells. BF (bright field), PE fluorescent (red), and BV421 fluorescent (blue) images were obtained. The scale bar is 20 μm. f An example of T-cell identification using a merged image. After image analysis, the image shows CD8+ and CD57+ T cells marked with the corresponding colors. The scale bar is 100 μm
2.1.2 Preparation of the PDMS Microfluidic Channel and PEG/biotin-PEG Coated Glass Substrate
PDMS microfluidic channels were fabricated by conventional soft lithography using SU-8 molds (Fig. 1a, step ① − ③). The microfluidic design includes flow channels for sample injection and cell capture and a vacuum line for the channel assembly. The vacuum line surrounds the flow channels, creating the negative pressure to hold the PDMS channel down to the substrate. The flow channels are composed of a capture chamber, an inlet/outlet and a resistance channel. The capture chamber consists of three parallel lanes; each lane has a width of 750 μm and a length of 5000 μm. The field of microscope view used for imaging is ~ 780 × 780 μm2, which covers the capture chamber with 15 sections. The channel height was chosen to be 25 μm. A lower channel height enables a larger chance for cells to interact and bind with the capture antibody; however, a channel height that is too small can lead to channel clogging or filtration of large cells. The average diameter of a T cell is 18 μm, and 25-μm-tall channels were found to be sufficient to enable a sample to flow without obstruction. The channel height can also affect the shear stress, an important parameter for affinity-based cell capture. We fixed the channel height and controlled the shear stress by varying the flow rate (flow speed).
The channel flow was driven by vacuum pumping connected to the outlet. The blood sample and reagents for antibody immobilization and fluorescent labeling were added to the inlet reservoir by pipetting or inserting tubes with pre-loaded fluids. The desired flow rates were achieved by adjusting the fluidic resistance of the microfluidic device. To control the overall fluidic resistance of the device, the resistance channel part, which has a much larger fluid resistance than the capture chamber, was designed and connected in series with the capture chamber. The resistance channels have a width of 25 μm and varying lengths of 15, 30, 45 and 60 mm (Fig. 1a and Fig. 2a). Assuming a viscosity of 1 mPa∙s, the calculated flow rates and the shear stress for each fluid resistance are 5.6, 2.8, 1.9, and 1.4 μl/min and 3.98, 1.99, 1.32, and 1.00 dyne/cm2, respectively (SI Table 1). When used for biosensing applications, the PDMS surface typically suffers from significantly large non-specific binding of biomolecules and cells. Red blood cells (RBCs) and CD8+ T cells can be non-specifically bound to the top of the PDMS chamber. To reduce the non-specific binding, the PDMS device was coated with bovine serum albumin (BSA) before use (Fig. 1a, step ④) [33].

Optimization of capture efficiency with varying shear stresses. a A parallel channel device with 4 different resistance channel lengths. b The purity of CD8+ T cells among the captured cells with varying fluidic resistance. c Number of captured T cells per image with varying fluidic resistances. d Bright-field and PE fluorescence images of captured T cells. The scale bar is 200 μm
The glass substrate was prepared with a PEG and biotin-PEG coating and stored at – 4 °C (Fig. 1b). While the biotin-PEG provides binding sites for neutravidin, the PEG provides passivation to inhibit the non-specific binding of biomolecules and non-target cells [29, 31, 34], which improves the specificity of the cell capture. The ratio of the PEG and biotin-PEG was chosen as 100:10 (SI Fig. 1). When comparing the ratios of biotin-PEG at 5:100, 10:100, and 20:100, it was observed that the 5:100 ratio captured the least number of T cells. As the ratio was increased, more T cells were captured, but there was not a significant difference between the results of 10:100 and 20:100. These ratios correspond to relatively large biotin-PEG concentrations compared to a typical biotin-PEG surface preparation protocol. An increase in biotin-PEG does not directly lead to an increase in the neutravidin coverage, because of the stoichiometry of biotin-neutravidin binding. With the large biotin coverage on the substrate, the corresponding neutravidin coverage falls in the plateau and is nearly at its maximum [35]. We expect this protocol to allow nearly the full coverage of neutravidin on the substrate.
2.1.3 Cell Capture and Fluorescent Cell Counting
The PDMS channel and the glass substrate can be prepared in advance and stored until use. They can be assembled easily and quickly before the capture and analysis. As described in the previous section, vacuum pumping was applied at the channel outlet to serve as the pressure source for driving the fluid flow and at the vacuum line to hold the PDMS channel onto the glass substrate. After the device assembly, anti-CD8 antibody immobilization, T-cell capture, washing, and fluorescent labeling were performed sequentially (Fig. 1c). For the immobilization of the antibody, neutravidin and biotinylated anti CD8 antibody were sequentially flowed through the capture chamber (Fig. 1c, step ① − ③). The incubation time was determined based on the binding kinetics of the fluorescein isothiocyanate (FITC)-biotin with neutravidin (SI Fig. 2). Whole blood or isolated WBC samples were injected into the device for the capture of the target T cells (Fig. 1c, step ④). Following the capture step, non-binding cells were washed with PBS, and fluorescent conjugated antibody cocktail was injected for counting of the specific T cells (Fig. 1c, step ⑤-⑥).
Figure 1d shows a microfluidic device setup on a microscope stage. A micropipette tip was used for the inlet reservoir. The tubing for the vacuum outlet and channel outlet was connected to a vacuum pump (DOA-P704-AC, Gast Manufacturing Inc.). After finishing the capture and labeling procedures, fluorescent and bright field microscopy images were taken at 9 positions in the capture chamber. Fluorescent images were processed and analyzed to quantify the CD57+CD8+ T cells among CD8+ T cells (Fig. 1e). CD8+ T cells were identified using a filter set for Phycoerythrin (PE) and CD57+ T cells for Brilliant Violet 421 (BV421). Counting the number of cells was done by applying an intensity threshold and particle counting with ImageJ. Unwanted cells can non-specifically adhere to the capture surface, but fluorescence analysis can easily eliminate them as they hardly fluoresce. Occasionally, non-specifically adherent non-CD8+ cells were identified as CD57+. A PE and BV421 overlay prevents those cells from being detected, and they were not included in the CD57+CD8+ T-cell counts. Bright-field images are not used to quantify the CD57+CD8+/CD8+ T-cell ratio, but they are used to optimize the capture process, especially the washing step. Comparison of the bright field and fluorescent microscopy images also enabled us to quantify non-specific binding. Figure 1f shows a typical image of captured CD8+ T cells after merging the bright field, PE and BV421 fluorescence microscopic images. Such images typically contain ~ 100 CD8+ T cells per field for the analysis. While the size of the capture chamber is large enough to obtain 15 images, 9 images (3 from each lane) were taken to minimize the influence of the inlet and the outlet. About 500 cells are sufficient to measure the CD57+CD8+/CD8+ T-cell ratio accurately.
2.2 Capture and Analysis Using Isolated WBCs
2.2.1 Optimization of the Capture Efficiency with Varying Shear Stresses
Shear stress has a critical role in affinity-based cell capture in microfluidic devices. The fluidic resistance in our microfluidic system determines the shear stress because the flow is driven by a fixed pressure difference of 1 atm. In practice, the fluidic resistance was controlled by the length of the resistance channels. Resistance channels with 15, 30, 45, and 60 mm lengths were tested to find the optimal flow speed (or shear stress). The calculated flow rate, flow speed, and shear stress for each device are summarized in Table S1. As shown in Fig. 2a, the capture devices with four different resistance channels are connected in parallel to conduct simultaneous tests. These four parallel test channels share the outlet at the center of the microfluidic device, while they have separate inlets for reagent and sample injections. In the device picture, the microfluidic capture channels were filled with red ink, and the vacuum line was filled with black ink for visualization.
Before using blood samples to capture CD8+ T cells, we tested our capture device with isolated WBCs. WBCs were enriched by density-gradient centrifugation from whole blood samples. Captured cells were imaged using bright field and fluorescent microscopy (Fig. 2d). As expected, the number of captured cells increased with the increasing channel length and lower shear stress. The purity of the CD8+ T cells among the captured cells was more than 98% in all cases, which proves the high selectivity of our capture method (Fig. 2b). In the device with a 15 mm resistance channel, an average of ~ 80 T cells were captured in one picture. In contrast, the yield gradually increased with longer resistance channels, with 186 and 174 T cells captured in devices with 45 and 60 mm resistance channels, respectively (Fig. 2c). We concluded that devices with 45 or 60 mm resistance channels provide suitable flow rates for CD8+ T-cell capture. High flow rates, or high shear stress, can detach weakly bound T cells with lower expression of CD8 molecules and reduce the number of captured T cells. Under conditions with the 45 and 60 mm fluidic resistance channels, the count of captured cells shows a plateau. We further tested if the frequency of CD57+ cells among captured T cells was consistent when the devices with 45 mm and 60 mm resistance channels were used. The CD57+CD8+/CD8+ T-cell ratio was measured consistently in the repeated experiments in both resistance channel designs (SI Fig. 3). These results suggest that the shear stress range of our capture device seems to have no significant impact on the detachment of captured T cells.

Comparison of the frequency of senescent CD57+CD8+ T cells using isolated WBCs. a The frequency of CD57+CD8+ T-cell subsets using multi-color flow cytometry. The subsets of lymphocytes were determined through sequential gating. b Measured values of the CD57+CD8+/CD8.+ T-cell ratio from the capture device compared with values from flow cytometry using isolated WBC samples. The different colors indicate samples obtained from different donors (N = 5)
2.2.2 Quantification of the CD57+CD8+/CD8+ T-Cell Ratio from Isolated WBC Samples
CD57+CD8+ and CD8+ T-cell counts were measured using microfluidic devices. A resistance channel of 45 mm in length was chosen for the tests. Isolated WBC samples were prepared from whole blood obtained by venipuncture from 5 healthy donors. Initially, the frequency of CD57+CD8+ T-cell subsets was determined using multi-color flow cytometry (Fig. 3a). WBCs stained with fluorochrome-conjugated monoclonal antibodies were gated based on their forward and side scatter properties and distinguished by their respective fluorescence intensities.
The CD57+CD8+/CD8+ T-cell ratios obtained from the microfluidic device were plotted against the flow cytometry results for each donor (Fig. 3b). The microfluidic measurements were performed at least 3 times per donor using different devices. The ratios were deduced from more than 500 captured T cells for each measurement. The CD57+CD8+/CD8+ T-cell ratios varied from approximately 14% to 32%, depending on the donors. The results show good agreement between the two quantification methods. This confirms that our microfluidic capture device efficiently captures CD8+ T cells, and the subsequent imaging analysis provides reliable measurements of the surface expression, yielding accurate CD57+CD8+ T-cell frequencies.
In principle, multi-color flow cytometry is superior to the microfluidic device for quantifying CD57+CD8+/CD8+ T-cell ratios because multi-color flow cytometry employs four fluorescence parameters, CD14/CD19, CD3, CD8, and CD57 for detecting CD57+CD8+ T cells whereas the microfluidic device relies on two parameters, CD8 and CD57. However, the microfluidic device was shown to be sufficient for accurate quantification of CD57+CD8+/CD8+ T-cell ratios. This can be explained by the fact that CD8 is almost exclusively expressed by CD3+CD14−CD19− T cells.
2.3 Capture and Analysis Using Whole Blood Samples
2.3.1 Optimization of the Capture Condition for Diluted Whole Blood
Under identical experimental conditions with isolated WBCs, it was observed that the capture efficiency of CD8+ T cells was significantly reduced when whole blood was utilized. This phenomenon can be primarily attributed to the RBCs inherent in whole blood, which are typically 1000 times more abundant than WBCs. RBCs in blood samples interfere with the physical interactions between the CD8+ T cells and the antibodies on the surface, often clogging the channel, especially at the inlet filter, resulting in a higher fluidic resistance. Moreover, clogged RBCs can exert a sweeping effect that dislodges captured T cells, thereby causing them to detach. Even though the dilution of the blood sample resulted in the capture of fewer T cells (SI Fig. 4), we opted to use diluted blood samples for the capture and further optimized the conditions to ensure the reproducibility of our results. Given that the quantity of WBCs traversing the capture chamber was crucial in enhancing the capture yield, we adjusted our sample injection protocol to enable a continuous flow of samples instead of a one-time chamber fill.

Optimization of capture conditions for the blood samples. a Fluorescent (PE) images of captured CD8.+ T cells with the injection/incubation and flowing conditions. Number of captured T cells (per image) depending on the capture method: injection/incubation or flowing conditions. b Number of captured T cells with varying dilution factors. c Non-specific binding on the top PDMS surfaces with varying dilution factors. The number of captured T cells with varying flow rates (fluidic resistances) d Number of captured T cells with varying resistance channels for a dilution factor of 2. e Number of captured T cells with varying resistance channels for a dilution factor of 4
The blood samples were flowed for 20 min instead of a 15 min incubation after the 5 min flow injection. For the × 2 diluted whole blood samples, the capture yield was improved ~ 2.5 times (Fig. 4a). With the new flow and capture protocol, we tested the capture yield with varying dilution factors of 2, 4, 8, and 16 (Fig. 4b–e). First, the decrease in the number of captured CD8+ T cells was confirmed again with the increase of the dilution factor (Fig. 4b). The 45 mm resistance channel was used here. Next, the degree of non-specific binding was investigated. The bottom substrate was passivated by PEG, where non-specific binding can barely be found. However, non-specific binding on the PDMS channel surface, passivated by BSA, could not be perfectly prevented. The WBCs bound on the top PDMS channel surface can be easily identified by unfocused cell images under bright field microscopy as these cells are located at different z-height. The number of non-specific bindings, represented by the top binding of the WBCs, was counted while controlling for the dilution factor (Fig. 4c). The non-specific binding seems to decrease with an increasing dilution factor. In most cases, the top binding ratios were small (~ 1 − 3%). In the case of the capture with × 2 diluted blood, dense clusters of top binding WBCs were observed in some images. Such non-specific binding events increased the average number of top binding WBCs and the variance of the data. Except in such cases, non-specifically bound WBCs on the top PDMS surface were less than 1 − 2%.
Quantifying the CD57+CD8+/CD8+ T-cell ratio requires capturing a sufficient number of CD8+ T cells. Assuming random sampling, a minimum of 100 cells is required, and > 500 cells are preferred to provide accurate measurements. In Fig. 4c, blood samples with × 2 and × 4 dilution factors provided a sufficiently large number of captured T cells. We further optimized the capture yield for T cells by adjusting the resistance channel for the × 2 and × 4 diluted blood samples (Fig. 4d and e). When the channel length was increased, the smaller shear rate increased the capture yield, while a lower flow rate, or fewer WBCs, did the opposite. These results suggest that compared to shear stress, the number of cells passing the chamber is the more dominant factor for T-cell capture under the given conditions. In the case of × 4 diluted blood, the increase in yield with the shorter resistance channel was less pronounced than the × 2 diluted blood.
The flow rate calculated for the isolated WBC samples cannot be used for whole and diluted blood samples because the viscosity of blood changes with the hematocrit [36]. For the × 2 diluted blood, the viscosity is expected to be ~ 3 times larger than the viscosity of water. For the capture devices with the 15 − 60 mm resistance channels, the flow rate changes from 1.56 to 0.39 μl/min. Similarly, for the × 4 diluted blood, the viscosity becomes ~ 2 times larger than the viscosity of water, and the flow rate varies from 2.07 to 0.52 μl/min. For the cell capture using isolated WBCs, the 45 mm resistance channel gave a sufficiently large capture yield, with a flow rate of 1.86 μl/min. In the case of whole blood samples, the × 4 diluted blood in the 15 mm resistance channel gives a similar flow rate (2.07 μl/min), which has the largest shear rate among the conditions shown in Fig. 4e. Given the exceptional reproducibility and elevated capture yield observed, our selection was to use blood samples diluted by a factor of four, in conjunction with 15 mm resistance channels, for subsequent measurements pertaining to the frequency of the CD57+CD8+ T-cell subset. With the given flow condition and the dilution factor, 10 µl of whole blood (before dilution) is injected in the capture device, which can contain ~ 10,000 WBCs. Note the population of CD8 + T cells is typically ~ 1/3 of WBCs and the image area is ~ half of the capture chamber area. Our capture device captures roughly ~ 10—20% of total CD8+ T-cell present in the sample.
2.3.2 Quantification of the CD57+CD8+/CD8+ T-Cell Ratio from Whole Blood Samples
Finally, we quantified the CD57+CD8+/CD8+ T-cell ratios from diluted blood using the conditions deduced in the previous section. The measured values using the microfluidic device were compared with the flow cytometry results (Fig. 5). Similar to the correlation observed with the isolated WBC samples shown in Fig. 3, the frequencies of the CD57+CD8+ T-cell subset in the whole blood samples correlated well between the data collected from the device and the flow cytometer. Our results confirm the accuracy of the CD57+CD8+ T-cell quantification using blood samples as small as a few tens of microliters, as it successfully captures the CD8+ T cells and enables fluorescence-based detection of CD57+CD8+ T cells.

Comparison of the frequency of senescent CD57+CD8+ T cells using whole blood. Measured values of the CD57+CD8+/CD8.+ T-cell ratio from the capture device compared with the values from flow cytometry using whole blood samples. Different colors indicate samples obtained from different donors (N = 5)
3 Conclusion
We developed a microfluidic device that enables rapid and straightforward quantification of the frequency of senescent CD57+CD8+ T cells from either isolated WBCs or diluted whole blood samples. The microfluidic device is composed of a biotin-PEG substrate and a PDMS microfluidic channel. The microfluidic flow was controlled by the resistance channel length and optimized for the efficient capture of CD8+ T cells. Captured CD8+ T cells were subsequently labeled with fluorochrome-conjugated anti-CD8 and anti-CD57 antibodies. This approach gives us an unbiased measurement of the frequency of CD57+CD8+ T cells, confirmed by comparing the results obtained from the microfluidic capture device with a flow cytometer.
The easy-to-perform analytical device presented here is anticipated to aid clinical practice and immunological research, where individual monitoring of senescent T-cell frequencies is crucial. Especially, the microfluidic method does not require user expertise and eliminates the need for preprocessing. In the field of immunology, confirming specific subsets of immune cells typically involves processing and analysis of samples using expensive equipment such as a flow cytometer by trained professionals, making the process time-consuming and high-cost. The microfluidic capture device allows a significant reduction of blood sample volume down to ~ 10 µl. Such a small amount of blood sample volume can be taken by fingerstick, which can give a huge advantage in applications of the technique by removing the requirement of blood sample collection by personnel with medical training. In conclusion, our technology would provide a convenient way to analyze surface molecule expressions, including but not limited to CD8 and CD57 molecules on cells, and help quantify specific immune cell subsets at high efficiencies.
4 Methods and Materials
4.1 PDMS Microfluidic Device Fabrication
For PDMS molds with a thickness of 25 µm, SU-8 3025 photoresist was photolithographically patterned using MA6 (Suss, MicroTec). PDMS (Sylgard 184) was mixed (base:curing agent = 1:10) and poured onto the mask mold. After curing at 65 °C for 2 h, the PDMS channel was peeled off from the mold, and the inlet and outlet holes were punched. The PDMS channel was coated with BSA by filling with BSA 5% solution and blow-drying with nitrogen gas.
4.2 Slide Glass PEGylation
Slide glasses were cleaned with piranha solution for 30 min and then rinsed with DI water and methanol for 10 min. Then, aminosilanization was performed on the glass slide using AEAPTMS (N-[3-(Trimethoxysilyl)propyl]ethylenediamine, Sigma-Aldrich) (Fig. 1b, step ②). For the saline reaction, the cleaned slide glasses were placed in a beaker containing a mixed solution of 100 mL of methanol, 5 mL of acetic acid, and 3 mL of AEAPTMS. The slide glasses were cleaned with methanol and dried using N2 gas. Subsequently, they were immediately placed in a moisture box. Next, 5 mL of DI water was poured into a tube containing 350 g of potassium sulfate and 45 mg of sodium bicarbonate, and the mixture was stirred. Then, 1 mL of a potassium sulfate and sodium bicarbonate solution was added to a tube containing 100 mg of mPEG-SVA (methoxy polyethylene glycol N-hydroxysuccinimidyl valerate, MW5000, Laysan Bio, Arab, AL) and 10 mg of biotin-PEG-SVA (biotinylated polyethylene glycol N-hydroxysuccinimidyl valerate, MW5000, Laysan Bio). This solution was filtered through a 200 nm filter. The filtered solution was sprayed onto one side of the slide glass and covered with another slide glass. The samples were then incubated for 2 h without exposure to light. A mixture of mPEG and biotin-PEG was reacted with the amine group resulting in a layer of PEG and biotin-PEG (Fig. 1b, step ③). After incubation, each pair of slide glasses was separated, washed with DI water, and dried with N2 gas. The slide glasses were individually placed into Falcon tubes filled with N2 gas.
4.3 Collection of Blood Samples and Enrichment of WBCs
Peripheral blood was obtained from healthy human donors in ethylenediaminetetraacetic acid tubes. No additional anticoagulation agent was used. WBCs were isolated by density-gradient centrifugation using a lymphocyte separation medium (density of 1.077–1.080 g/mL; Corning Inc.). After isolation, cells were cryopreserved in fetal bovine serum (Corning Inc.) with 10% dimethyl sulfoxide (Sigma-Aldrich) at – 180 °C until use. This study was approved by the Institutional Review Board at the Korea Advanced Institute of Science and Technology (IRB, No. KH2023-018), and all subjects provided informed consent.
4.4 Cell Capture Procedure
For the immobilization of the antibody, first, neutravidin (0.3 mg/ml, Thermo Fisher Scientific) was filled in the capture chamber and incubated for 1 min (Fig. 1c, step ②). A sufficiently large concentration of neutravidin was used for quick binding and maximal coverage. Then, the biotinylated anti-CD8 antibody (25 µg/ml, 555,365, BD Pharmingen) was filled in the chamber and incubated for 10 min (Fig. 1c, step ③). The incubation time was determined based on the binding kinetics of the fluorescein isothiocyanate (FITC)-biotin with neutravidin (SI Fig. 2). Once the antibody was immobilized and the capture device was ready, samples, either whole blood or isolated WBCs, were injected into the chamber (Fig. 1c, step ④). The samples were introduced in the capture chamber (continuously flowed or incubated after injection) for capturing the target cells, and the non-target cells were washed out with phosphate-buffered saline (PBS) for 10 min (Fig. 1c, step ⑤).
4.5 Fluorescence Imaging Analysis
After capturing CD8+ T cells in the capture chamber, CD8+ and CD57+ T cells were labeled with fluorochrome-conjugated antibodies. PE and BV421-conjugated antibodies were used to label the CD8+ and CD57+ T cells (555,367, BD Pharmingen and 563,896, BD Horizon). Solutions were prepared by mixing 40 μl of PE-conjugated anti-CD8 antibody (555,367, BD Pharmingen) and 20 μl of BV421-conjugated anti-CD57 antibody (563,896, BD Horizon) to 140 μl of PBS. The solution was stored in a refrigerator until ready to use. A PE anti-CD8 and BV421 anti-CD57 antibody cocktail was injected and incubated for 10 min (Fig. 1c, step ⑥).
Images of 780 × 780 μm in size were captured from 9 locations per device. A bright field image and two types of fluorescence images (PE, BV421) were obtained for each location. The following filter settings were used to obtain the fluorescence images of PE (EX 540/25, DM 565, BA 605/55) and BV421 (BV-421: EX 389/38, DM 414, BA 433/24). A threshold method was applied to discriminate the background signal in each fluorescence image. PE (anti-CD8) used the yen method, and BV421 (anti-CD57) used the triangle method. These methods analyzed the intensity distribution over the entire image area and determined the threshold for signal detection. The thresholding process was similar to gating procedures on a flow cytometer and helped distinguish positive signals from the background. The threshold mask was merged with the fluorescence image to confirm the expression of CD8 and CD57 molecules on the cells (Fig. 1e). This step involved overlapping the fluorescence images with a mask that identified signal regions. First, the number of CD8+ T cells was counted in the PE positive area determined by the Yen thresholding method with the particle analysis function of ImageJ. Similarly, to count the number of CD57+ T cells, the BV421 fluorescence image was used to determine the BV421 positive area using the triangle method. By overlapping the BV421 positive area and the PE positive area, the number of CD57+CD8+ T cells was calculated using the particle analysis function only for the areas where both are positive (Fig. 1f).
4.6 Flow Cytometric Assessment
Multi-color flow cytometry was used to evaluate the frequency of CD57+CD8+ T cells among CD8+ T cells using blood samples. After thawing, the cells were washed with PBS and stained with LIVE/DEAD-Aqua (Invitrogen) for 20 min at room temperature, followed by a cocktail of surface antibodies for 20 min at room temperature, including BV510 anti-CD14, BV510 anti-CD19, BV650 anti-CD4, BV786 anti-CD3, APC-Cy7 anti-CD8 (BD Biosciences) and PE-Cy7 anti-CD57 (Invitrogen). Upon completion of staining, cells were analyzed on an LSR II instrument (BD Biosciences), and the data was analyzed using the FlowJo software version 10.5.3 (FlowJo, LLC.).
Data availability
The datasets used and/or analysed during the current study available from the corresponding authors on reasonable request.
References
Covre, L.P., De Maeyer, R.P.H., Gomes, D.C.O., Akbar, A.N.: The role of senescent T cells in immunopathology. Aging Cell 19(12), 1–9 (2020)
Weekes, M.P., Wills, M.R., Mynard, K., Hicks, R., Sissons, J.G.P., Carmichael, A.J.: Large clonal expansions of human virus-specific memory cytotoxic T lymphocytes within the CD57+ CD28- CD8+ T-cell population. Immunology 98(3), 443–449 (1999)
Brenchley, J.M., Karandikar, N.J., Betts, M.R., Ambrozak, D.R., Hill, B.J., Crotty, L.E., Koup, R.A.: Expression of CD57 defines replicative senescence and antigen-induced apoptotic death of CD8+ T cells. Blood 101(7), 2711–2720 (2003)
Furman, D., et al.: Chronic inflammation in the etiology of disease across the life span. Nat. Med. 25(12), 1822–1832 (2019)
Strioga, M., Pasukoniene, V., Characiejus, D.: CD8+CD28- and CD8+CD57+ T cells and their role in health and disease. Immunology 134(1), 17–32 (2011)
Youn, J.C., et al.: Immunosenescent CD8+ T cells and C-X-C chemokine receptor type 3 chemokines are increased in human hypertension. Hypertension 62(1), 126–133 (2013)
Tae Yu, H., et al.: Characterization of CD8+ CD57+ T cells in patients with acute myocardial infarction. Cell. Mol. Immunol 12(4), 466–473 (2015)
Lee, Y., et al.: Senescent T cells predict the development of hyperglycemia in humans. Diabetes 68(1), 156–162 (2019)
Alpert, A., et al.: A clinically meaningful metric of immune age derived from high-dimensional longitudinal monitoring. Nat. Med. 25(3), 487–495 (2019)
Cossarizza, A., et al.: Guidelines for the use of flow cytometry and cell sorting in immunological studies (third edition). Eur. J. Immunol. 51(12), 2708–3145 (2021)
Vergara-Barberán, M., Carrasco-Correa, E.J., Lerma-García, M.J., Simó-Alfonso, E.F., Herrero-Martínez, J.M.: Current trends in affinity-based monoliths in microextraction approaches: a review. Anal. Chim. Acta 1084, 1–20 (2019)
Perez-Gonzalez, V.H., Gallo-Villanueva, R.C., Camacho-Leon, S., Gomez-Quiñones, J.I., Rodriguez-Delgado, J.M., Martinez-Chapa, S.O.: Emerging microfluidic devices for cancer cells/biomarkers manipulation and detection. IET Nanobiotechnol. 10(5), 263–275 (2016)
Nahavandi, S., et al.: Microfluidic platforms for biomarker analysis. Lab Chip 14(9), 1496–1514 (2014)
Roh, S., Jang, Y., Yoo, J., Seong, H.: Surface modification strategies for biomedical applications: enhancing cell-biomaterial interfaces and biochip performances. Biochip J. 17(2), 174–191 (2023)
Son, M.H., Park, S.W., Sagong, H.Y., Jung, Y.K.: Recent advances in electrochemical and optical biosensors for cancer biomarker detection. Biochip J. 17(1), 44–67 (2022)
Li, P., Gao, Y., Pappas, D.: Negative enrichment of target cells by microfluidic affinity chromatography. Anal. Chem. 83(20), 7863–7869 (2011)
El-Ali, J., Sorger, P.K., Jensen, K.F.: Cells on chips. Nature 442, 403–411 (2006)
Hou, S., et al.: Polymer nanofiber-embedded microchips for detection, isolation, and molecular analysis of single circulating melanoma cells. Angew. Chemie - Int. Ed. 125(12), 3463–3467 (2013)
Neoh, K.H., et al.: PH-responsive carbon nanotube film-based microfluidic chip for efficient capture and release of cancer cells. ACS Appl. Nano Mater. 5(5), 6911–6924 (2022)
Murthy, S.K., Sin, A., Tompkins, R.G., Toner, M.: Effect of flow and surface conditions on human lymphocyte isolation using microfluidic chambers. Langmuir 20(26), 11649–11655 (2004)
Nagrath, S., et al.: Isolation of rare circulating tumour cells in cancer patients by microchip technology. Nature 450, 1235–1239 (2007)
Gleghorn, J.P., et al.: Capture of circulating tumor cells from whole blood of prostate cancer patients using geometrically enhanced differential immunocapture (GEDI) and a prostate-specific antibody. Lab Chip 10(1), 27–29 (2010)
Yan, S., et al.: Integrated multifunctional electrochemistry microchip for highly efficient capture, release, lysis, and analysis of circulating tumor cells. Anal. Chem. 89(22), 12039–12044 (2017)
Lu, Y.T., et al.: NanoVelcro chip for CTC enumeration in prostate cancer patients. Methods 64(2), 144–152 (2013)
Murlidhar, V., et al.: A radial flow microfluidic device for ultra-high-throughput affinity-based isolation of circulating tumor cells. Small 10(23), 4895–4904 (2014)
Stott, S.L., et al.: Isolation of circulating tumor cells using a microvortex-generating herringbone-chip. Proc. Natl. Acad. Sci. U. S. A. 107(43), 18392–18397 (2010)
Deliorman, M., et al.: AFM-compatible microfluidic platform for affinity-based capture and nanomechanical characterization of circulating tumor cells. Microsyst. Nanoeng. 6(1), 20 (2020)
D’souza, A.A., Shegokar, R.: Polyethylene glycol (PEG): a versatile polymer for pharmaceutical applications. Expert Opin. Drug Deliv. 13(9), 1257–1275 (2016)
Abdallah, B. G., Ros, A.: Surface coatings for microfluidic-based biomedical devices. Woodhead Publishing Limited (2013)
Hong, S., et al.: Covalent immobilization of selectins enhances cell rolling. Langmuir 23(24), 12261–12268 (2007)
Misiakos, K., Kakabakos, S.E., Petrou, P.S., Ruf, H.H.: A monolithic silicon optoelectronic transducer as a real-time affinity biosensor. Anal. Chem. 76(5), 1366–1373 (2004)
Ryu, J. Y., et al.: Profiling protein–protein interactions of single cancer cells with in situ lysis and co-immunoprecipitation. Lab Chip 19(11), 1922–1928 (2019)
Ma, G.J., Ferhan, A.R., Jackman, J.A., Cho, N.J.: Conformational flexibility of fatty acid-free bovine serum albumin proteins enables superior antifouling coatings. Commun. Mater. 1, 45 (2020)
Zhang, Z., Feng, X., Xu, F., Hu, X., Li, P., Liu, B.F.: An improved surface modification of poly(dimethylsiloxane) via a green chemistry approach for on-chip biomolecular separation. Anal. Methods 5(18), 4694–4700 (2013)
Hamming, P.H.E., Huskens, J.: Streptavidin coverage on biotinylated surfaces. ACS Appl. Mater. Interfaces 13(48), 58114–58123 (2021)
Trejo-Soto, C., Hernández-Machado, A.: Normalization of blood viscosity according to the hematocrit and the shear rate. Micromachines 13(3), 1–20 (2022)
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (NRF-2021R1A2C1005807), and by Samsung Science and Technology Foundation (SSTF-BA1802-05).
Funding
Open Access funding enabled and organized by KAIST.
Author information
Authors and Affiliations
Contributions
Study design: Y.-H.C., D. L., E.-C. S. and W. L. Experiment and data collection: Y.-H.C., W.-J.K., D.L. and B.-J.J. Data analysis and interpretation: Y.-H.C., W.-J.K., E.-C.S. and W.L. Writing: Y-H.C., W.-J.K., E.-C.S. and W.L. with comments from all authors.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Choi, Yh., Kim, WJ., Lee, D. et al. Microfluidic Capture Device for Simple, Cell Surface Marker-Based Quantification of Senescent CD8+ T Cells. BioChip J (2024). https://doi.org/10.1007/s13206-024-00142-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13206-024-00142-6