
Abstract
Corrosion processes were studied in epigenic caves of the Moravian Karst (MK). Samples of naturally corroded speleothems and carbonate rocks collected in various MK caves were compared with limestone and calcite standards that were corroded under well-controlled conditions in the laboratory. Two stages of corrosion were distinguished: (1) an early stage of pitting and linear corrosion predisposed by point and line defects in the standards, and (2) an advanced stage characterized by deep corrosion of the entire standard surface. During the second stage, the internal structure of the corroded substrate remained partially preserved, however, if the water was extremely aggressive, the corrosion progressed to depth en-bloc regardless of the structure. In this paper, we (1) proposed a new corrosion mechanism based on the binding of aqueous calcium by clays and tested it experimentally in the cave, (2) characterized conditions that generally favor the formation of aggressive waters (water undersaturated with calcite) into seven categories incorporating them into a conceptual model of the karst vertical profile, and (3) discussed anthropogenic influences on these processes.
Similar content being viewed by others

Introduction
Corrosion of calcite speleothems is a current environmental problem in Moravian Karst caves (e.g., straw stalactite falling). In general, it is damage caused to secondary solid carbonates via dissolution by aggressive solutions (water undersaturated by carbonates). A question of concern is whether human activities participate in this process and, if so, how to mitigate this impact.
Carbonate dissolution, basic corrosion process, has been intensively studied during the last five decades, mainly in the calcite–water–CO2 system (Plummer et al. 1978; Dreybrodt 1981; Rickard and Sjoberg 1983; Busenberg and Plummer 1986; Chou et al. 1989; Schott et al. 1989; Morse et al. 2002; Arvidson et al. 2003; Xu et al. 2012; Peng et al. 2015; Subhas et al. 2015; Veetil et al. 2018; Carrasco et al. 2021). However, it is necessary to study the corrosion conditions separately for hypogenic and epigenic caves. While hypogenic caves are associated with special conditions such as elevated temperatures with endogenous fluids emerging from below (hydrothermal water, elevated CO2 levels, sulphane, sulfuric acid, etc., see Audra et al. 2010; Plan et al. 2012; Klimchouk et al. 2017; Frumkin et al. 2018; Columbu et al. 2021; Laurent et al. 2021, 2023), epigenic caves are formed by shallow circulation of meteoric water (see, e.g., Palmer 1991; Audra and Palmer 2011; or Laurent et al. 2021).
Current examples of corrosion in epigenic caves can be classified according to mechanism as being either non-specified (Audra and Palmer 2011; Sala and Bella 2023), meteoric impact (Martín-García et al. 2011), condensation corrosion (Cailhol et al. 2019; Dublyansky and Spötl 2015), dripwater impact (Kukuljan et al. 2021), or metals impact (Gázquez et al. 2013). Corrosion by bat guano is discussed in Audra et al. (2019, 2021) and Sala et al. (2023). A very specific example of mechanical corrosion/abrasion by blast furnace slag was recently published by Faimon et al. (2022a, b).
This work is focused primarily on epigenic caves of the Moravian Karst. Despite many studies worldwide, the causes and conditions of corrosion are still not fully understood. Because the corrosion process is very difficult to study in situ, mainly due to slow dynamics and lack of tools to monitor the development of the corrosion over time, we supported this study with laboratory experiments. The objectives of this study were (1) to compare the samples of naturally corroded limestone/calcite with the standards artificially corroded under well-controlled laboratory conditions, (2) to summarize the known and less known corrosion mechanisms and conditions controlling this process, (3) to discuss them using a conceptual vertical karst profile model, (4) to present new mechanisms based on side reaction, and (5) to estimate anthropogenic influence.
Site of the study
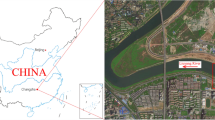
The study was carried out in the Moravian Karst (MK), the most extensive karst area in the Czech Republic (Fig. 1). It is located north-east of Brno (49°13´ to 49°25´ N, 16°38´ to 16°47´ E) in a part of the Drahany Highlands. The elevation of the karst plateau varies between 250 and 550 m above sea level, forming a zone 3 to 5 km wide and 25 km long and covering an area of about 94 km2 (Blecha and Faimon 2014). Granitoid rocks of the Brno massif (proterozoic age) form the basement. In addition to some Jurassic sandstones/limestones and Cretaceous sediments (Rudice beds), MK is mainly formed by middle Devonian to lower Mississippian limestones, which were deposited in two main formations: (1) older Macocha Fm. made up of more than 1000 m of cyclic reef (Vilémovice Lst.) and lagoonal (Lažánky Lst.) limestone and dolomite deposits (Eifelian–Frasnian) and (2) younger Líšeň Fm. comprising calciturbidites of the Hády-Říčka Lst. and more shallow water Křtiny Lst. (Musil et al. 1993). This mostly limestone sequence is overthrust by a Variscan flysch stack (the so-called Culmian facies). More than 1900 caves are known in MK, five of which are open to the public (Balcarka, Kateřinská, Punkevní, Sloup-Šošůvka, and Výpustek caves).

Site of the study. Geological map of the Moravian Karst indicating locations of caves where speleothems were sampled
The caves in which the speleothems were sampled were Amatérská cave, Harbešská cave, Řečiště cave, 17-Průtoková cave, Kateřinská cave, Punkevní cave, Nová Drátenická cave, Salmův Výpustek cave, Podmůstková cave, Holštejnská cave, T4 Sinkhole “u Kříže”, Vilémovice river sink, P1 Holštejn cave, and Pustožblebská cave. Additional speleothems were sampled for comparison in Mladeč caves, Javoříčko caves (Czech Republic), and Gombasek and Domica-Baradla cave systems (Slovakia/Hungary); see Table 1 for the cave characteristics.
Based on local hydrology, MK can be divided into northern, central, and southern parts, drained by the Punkva River, Jedovnický brook, and Říčka River, respectively. With an annual average temperature of 8.3 °C and an annual rainfall of 543 mm, MK belongs to a moderately warm and moderately humid climatic region with some drier and warmer sites in its southern part. Maximum precipitation occurs from July to August and minimum from February to March (see the climate classification of Kottek et al. 2006).
Theoretical background
The fundamental processes in carbonate karst are controlled by kinetics at (1) the water–air boundary,
and (2) the water–CaCO3(s) boundary,
where kf(1), kb(1), kf(2), and kb(2) are rate constants. Based on the principle of detailed balancing, the fraction of the rate constant for forward reaction and backward reaction is the equilibrium constant, kf(1)/kb(1) = KH (Henry’s constant) and kf(2)/kb(2) = KS (solubility product). Both processes expressed by Eqs. (1) and (2) are relatively slow and can often be found out of equilibrium under karst conditions.
In addition to kinetics on interface boundaries, the karst processes are controlled by equilibria in bulk solution
The dissociation processes covered in Eqs. (3) and (4) belong to the fastest ones. They quickly reach equilibrium and can always be assumed to be in equilibrium. The equilibrium constants for the processes described by Eqs. (3) and (4) are K1 and K2−1, respectively. Other dissolved components involved in the overall balance, such as hydroxy- and carbonate complexes, are less important (e.g., Stumm and Morgan 1996, or manual for PHREEQC, Parkhurst and Appelo 2013). Formally, Eqs. (1), (2), (3), and (4) can be combined, giving the overall equation for carbonate dissolution (from left to right) and calcite precipitation (from right to left) in an open system:
The rate constants, kf(s) and kb(s), quantify the fluxes from calcite to solution and back from solution to calcite. The equilibrium constant of the process is Kcc = kf(s)/kb(s). Alternatively, this constant may be obtained by combining the equilibrium Eqs. (1), (2), (3), (4) and relevant constants: KCC = KS K2−1 K1 KH.
Equilibria/disequilibria
The equilibrium of the process at the carbonate/water boundary given by Eq. (2) is quantified by the equation
where Ks is the equilibrium constant. Brackets indicate activities (the activity of solid carbonate is one and constant). Outside the equilibrium, the activity product, Eq. (6), is different from Ks:
where Qs is the reaction quotient (ion activity product, IAP, in other words).
The ratio Qs/Ks is saturation state Ω,
More frequently, saturation is expressed with the saturation index SI(calcite):
Negative SI(calcite) values mean undersaturation, whereas positive values indicate supersaturation. The solution is in equilibrium with calcite when SI = 0 (e.g., Appelo and Postma 2005).
The reaction quotient (ion activity product, IAP) of Eq. (5) is
where the brackets denote activity, and PCO2 is the partial pressure of gaseous CO2. Solid carbonate and water in Eq. (5) are pure and almost pure components, respectively, so their activities are assumed to be equal to unity and constant during the reaction. If QCC = KCC, the process (Eq. 5) is at equilibrium. Equation 10 indicates that the total mineralization of saturated water is controlled by \(P_{\text{CO}_{2g}}\) in the open system.
Dissolution mechanism
The dissolution mechanism of carbonate minerals can be derived from the three parallel processes proposed by Plummer LN, Wigley TML, and Parkhurst DL for dissolution kinetics (see the so-called PWP equation, Plummer et al. 1978). Originally, the PWP equations did not meet the principle of detailed balancing and hence were modified by Busenberg and Plummer (1986) and Chou et al. (1989) to the following form:
and
Equation 13 is also presented in the form
Equations (11), (12), (13), and (14) thus represent the three parallel processes, during which the carbonate surface is attacked by H+ ions (Eq. (11)), carbonic acid (Eq. (12)), and water (Eq. (14)). Note that no individual rate equation could be truncated by conjoint components, as is common practice in expressing equilibrium equations.
The mechanism of CO2 dissolution in water is most often described using the two-layer model of the interface CO2(g)–H2O; see Liss and Slater (1974) or Faimon et al. (2022a, b).
Methods
Speleothem and limestone samples
In total, we collected 162 samples of speleothems (mainly straws, plus small stalactites and fragments of curtains) from caves of the Moravian Karst and as well as more distant Czech Republic and Slovakia/Hungary caves, see Table 1 for detailed locations. Speleothems from Slovakia/Hungary caves, known for their high purity without corrosion, were sampled for comparison. The speleothems were cleaned in an ultrasonic bath and carefully sorted by the degree of corrosion. Subsequently, the selected samples were studied using the methods below. Corroded limestones were sampled from the Pršavý komín wall and the entrance passage (Býčí skála cave, MK) for detail analysis.
Limestone/calcite standards
Plates 2 × 3 × 0.5 cm in size were cut from the Lažánky Lst. (Býčí skála, MK), and subsequently ground, polished, and used as limestone standards in the experiments. Thin section images indicating both micrite and sparite components are presented in Supplementary Appendix A1. Natural calcite crystals (vein calcite, MK) were split along the cleavage planes into rhombohedrons approximately 1 × 1 × 1 cm in size that were used as calcite standards.
Laboratory experiments
The main aim of the experiments was to simulate corrosion under well-defined conditions and compare results with a manifestation of natural corrosion. The limestone and calcite standards were left to corrode in (a) 0.1 mol L−1 HCl for 3 days, (b) 5% acetic acid for 7 days, and (c) water saturated with CO2(g) at pressure P = 1.7 atm (0.0909 mol L−1) under conditions of a closed system for 7 and 20 days.
Field experiment
The limestone standards were carefully inserted into selected moist clays in the environment of the Býčí skála cave (MK) and left to corrode under cave conditions for 6 months. The mineral composition of the clays is in Table 2; diffractograms are shown in Supplementary Appendix A2.
Hydrogeochemistry
Two dripwater data sets from Moravian Karst were used to assess proposed corrosion conditions. Both data sets were analyzed with the same methodology—electrochemical properties were measured with a hand multimeter Greisinger G7500, the elemental composition was analyzed via ICP-MS (The Environmental Laboratory of the Transport Research Centre, Brno), carbonate alkalinity was analyzed via acidimetric titration (in the analytical laboratory of the Dept. of Geological Sciences, Masaryk University, Brno), and saturation index to calcite was calculated using PHREEQC with the default database Parkhurst and Appelo (2013). The drip water discharges were determined by (1) counting drops at water sampling in Amatérská cave and (2) measured continuously using a custom-made logger in Býčí skála cave.
Laboratory techniques and equipment
Scanning electron microscopy (JEOL JSM-6490 LV with an SEM–EDX microanalyzer (INCAx-act, Oxford Instruments) was used to study the gold-plated surfaces of both naturally corroded and experimentally corroded limestone / calcite standards using an accelerating voltage of 15 kV. All surfaces were scanned linearly in detail. The analytical signal was processed using the simplified XPP matrix correction routine (Pouchou and Pichoir 1991).
Binocular microscope Olympus SZX16 coupled with an Olympus E620 DSLR camera was used for the optical study of sample surfaces (artificially and naturally corroded limestone/calcite), and a polarizing microscope (Nikon Eclipse ME 600) was used for the study of polished thin sections.
The bulk phase compositions of clays / muds were determined by powder X-ray diffraction (XRD). Analysis was carried out using a Panalytical X'Pert PRO MPD diffractometer with reflection geometry equipped with a cobalt tube (λKα = 0.17903 nm), a Fe filter, and a 1-D RTMS detector (X'Celerator). The acquired data were processed using Malvern Panalytical HighScore 4 plus and Bruker AXS Topas 4 software. Quantitative phase analysis was performed using the Rietveld method (Rietveld 1969). The associations of clay minerals were identified by analyzing oriented mounts. The samples were gently ground with mortar and pestle, dispersed in distilled water, and subjected to repeated ultrasonic disintegration and centrifugation. A fraction with < 2 μm grain size was obtained in suspension by 750 rpm centrifugation for 3.3 min. Oriented mounts were prepared by depositing the dispersed clay particles onto glass slides and allowing the suspension to dry. The XRD patterns of each sample were recorded three times: air-dried, ethylene glycol solvated, and heated to 400 °C for 30 min., see diffractograms in Supplementary Appendix A2.
Unless otherwise stated, all analyses were carried out in the laboratories of Dept. of Geological Sciences, Faculty of Science at Masaryk University, Brno.
Results
Calcite speleothems
Calcite fabrics in speleothems were well described by Frisia (2015) or Frisia et al. (2018). This work provides supplemental information on the peculiar structure of straw stalactites (soda straw). The walls of a pristine soda straw are built from three layers, A, B, and C (see the sketch in Supplementary Appendix 3a), the middle of which (B) is load bearing (Faimon et al. 2006). The outer layer A of the straw is highly porous, probably formed by deposition of aerosols, dust particles, and other impurities (see, Fig. 2c and the analysis in Supplementary Appendix A4). The clearest glass-like straws are formed only by layer B (Supplementary Appendices A3b, c). In this case, the outer side of layer B is compact without pores (Supplementary Appendix A3d), although a specific structure is visible at higher magnification (Supplementary Appendix A3e). The inner side of B is made up of single radially arranged crystals of tabular calcite (Supplementary Appendix A3f and g) to feather-like calcite (Supplementary Appendix A3h–i). The inner layer C is formed by tiny individual crystals of secondary calcite that grow on the wall of the central channel (Fig. 2d). Soda straw stalactites and curtains are speleothems that are more prone to corrosion in epigenic caves of the Moravian Karst (Supplementary Appendix A5-1). Although stalagmites and flowstones are the dominant speleothems by weight, signs of corrosion are generally less noticeable, with some exceptions (Supplementary Appendices A5-2). The corrosion of speleothems could be supported by contamination with clays/muds (Supplementary Appendix A5-3).

Naturally corroded soda straws (Bludiště, Amatérská Cave, MK). Corrosion coupled with contamination of the outer surface of the straw (a, b, c); corrosion coupled with secondary calcite precipitated in the inner channel of the straw (d, e, f); corrosion of the outer walls of straws, (g, h); see the arrows
Corrosion
Examples of corroded soda straws collected in the Moravian Karst caves are shown in Fig. 2. The corrosion unsurprisingly propagates inwards from the surface. Corrosion of the outer surface of the soda straws is shown in Fig. 2e, f, g, and h. Sometimes, the straw channel is blocked by calcite growth when the seepage water is episodically supersaturated. Corrosion associated with precipitated secondary calcite in the inner channel of the straw is shown in Fig. 2d, f, and g. SEM micrographs of the outer surface of corroded straws show both positive (Fig. 3a) and negative relief (Fig. 3b). In some cases, the outer surface of the soda straw shows rather apparent porosity formed by a quantity of individual secondary calcite crystals (Fig. 3c), some of which are directionally arranged (Fig. 3d). SEM micrographs of the inner surface of corroded straws show complex patterns of tabular calcite, spatially limited (Fig. 4a and b) to scalenohedral calcite crystals (“dog-tooth-spar”) (Fig. 4c and d). The pattern of densely distributed scalenohedral calcite crystals is interrupted in places by deep depressions.

Naturally corroded soda straw (Bludiště, Amatérská cave, MK); details of the outer surface. Porous / contaminated surface (a, b), see highlighted ovals; apparent porosity formed by prismatic calcite crystals, formed by secondary precipitation (c, d), see the arrows showing a “directional growth”. SEM micrographs

Naturally corroded soda straw: the inner surface (Bludiště, Amatérská cave, MK). Tabular calcite, limited by the wall of the inner channel of straw (a, b) and scalenohedral calcite (dog-tooth calcite) (c, d). SEM images
Extreme corrosion of limestones was documented in the “Pršavý komín” (Býčí skála cave, MK), see Fig. 5. A general view of the weathered surface is in Fig. 5a; deep depressions formed by micrite dissolution (see hollow arrows) are in Fig. 5b. Enclaves of randomly oriented scalenohedral calcite crystals commonly resist corrosion (Fig. 5c and d, white arrows), whereas the carbonate cement between these enclaves is dissolved (Fig. 5d, hollow arrows). In Fig. 5e and f, signs of recrystallization (white arrows) and dog-tooth-spar calcite (hollow arrows) are visible. Corroded straws (above) and curtains (below) photographed in situ in the Amatérská cave (MK) are shown in Supplementary Appendix A5-1. Corroded limestone and flowstone in the Řečiště cave (MK) are shown in Supplementary Appendix A5-2. Corrosion coupled with mud contamination is shown in Supplementary Appendix A5-3.

Naturally corroded limestone (Pršavý komín, Býčí skála cave, MK). Total view (a); deep depressions created by micrite dissolution – hollow arrows (b) and (d), sparite enclave – white arrows (c, d); signs of calcite recrystallization – white arrows (e, f); dog-tooth calcite – hollow arrows (f). SEM images. See the text for details
Artificially corroded calcite/limestone standards allowed a comparison with naturally corroded speleothems, specifying the corrosion mechanisms: in the early stages of interaction with aggressive solutions, etched pits (Fig. 6a and b, hollow arrows) and longitudinal grooves (Fig. 6a, white arrows) were formed on the surface of calcite standards. During more advanced stages, continued corrosion exposed the inner structure of the limestone and calcite with well-contoured rhomboscalenohedral, scalenorhombohedral, to scalenohedral calcite crystals (Fig. 7a–f and Fig. 8d–e), respectively. In contrast, extremely aggressive waters corroded the standards en-bloc (compare the original surface of the standard in Supplementary Appendix A6a, b with the corroded surface in Supplementary Appendix A6c and d), and did not follow any preexisting crystal structure.

Artificially corroded calcite standards after a 7-day interaction with aggressive water (dissolved CO2 ~ 0.0909 mol L−1). Etched pits (see hollow arrows) (a, b); longitudinal grooves (see white arrows) (a). SEM images

Artificially corroded limestone standards after a 20-day interaction with aggressive water (dissolved CO2 ~ 0.0909 mol L−1): Corroded surface with a part of the original polished surface (left) (a) and (b); detailed morphology of the corroded surface with sparite enclaves of different orientations (see the circled details in the picture) (c–e); detailed view of scalenohedral calcite (dog-tooth-spar) (f). SEM images

Artificially corroded calcite standard after 7-day interaction with 5% acetic acid: fresh calcite standard (photo by J. Faimon) (a); detailed morphology of the fresh surface, see arrows indicating crystal layer steps (b, c); corroded calcite surface with uniform scalenohedral calcite (dog tooth calcite) (d, e). SEM images
Two sets of hydrogeochemical data from Amatérská cave and Býčí skála cave (both Moravian Karst) are presented in Table 3 to support knowledge on conditions of corrosion. Data were taken from Pracný et al. (2022). Both dripwaters show low electrical conductivity and low saturation index with deeply negative bottom values. Discharge of the drip water from the Zemní pyramidy dome (Amatérská cave, MK) shows extremely high flow variability. In contrast, the discharge of dripwater from the Pršavý komín (Býčí skála cave, MK) shows an extremely high flow with lower variability.
Based on empirical observations that calcite speleothems and carbonate rocks are more corroded in contact with clays (see Supplementary Appendix A5-3), the interaction of limestone standards with clays was tested in the environment of Býčí skála cave (MK). Results of these experiments are presented in Fig. 9. The limestone standard inserted in VS-clay showed extensive pitting corrosion (Fig. 9a and b), whereas the standards embedded in the VS-clayey-eluvium showed advanced corrosion with exposed tabular calcite (Fig. 9c, d, and e). Only weak signs of pitting corrosion were observed on the limestone standard imputed in the SS-Fe-clay (Fig. 9f).

Artificially corroded limestone standards after 6 months of contact with clay in the cave environment (Býčí skála cave, MK): Local corrosion of the standard in VS-clay, see the initial corrosion around limestone pores (a, b); the standard corroded in VS-clayey-eluvium, see the highlights in pictures (c–e); limestone standard after interaction with SS-Fe-clay, see highlights, showing signs of corrosion (f). VS-clay (a, b), VS-clayey-eluvium (c–e), and SS-Fe-clay (f)
Discussion
The natural morphology of soda straws predetermines, to some extent, their susceptibility to corrosion and other destructive processes. The porous outer layer A (Figs. 2c, 3a and b) retains water, prolonging the calcite–water interaction period and making the surface susceptible to corrosion (Fig. 2h). Airborne contaminants deposited on the surface could interact with this process. The lack of published works on this topic suggests that this process is clearly understudied. Layer B also supports corrosion due to its complicated structure. The outer diameter of a soda straw is generally defined by the diameter of a water drop, which is primarily controlled by the surface tension of water (see, e.g., Kalová and Mareš 2015) and, to some extent, by the flow rate (Genty and Deflandre 1998). The inner diameter of the soda straw is limited by a diffusive thickness of the water layer on the hanging drop that has degassed and supersaturated with respect to calcite (according to Eq. 5 from right to left, for more information see, e.g., Dreybrodt and Fohlmeister 2022). Note that the lifetime of the hanging drop (controlled by the discharge of the drip site) thus defines the thickness of the straw wall. Newly growing calcite crystals must arrange themselves in this very spatially limited cylindrical wall (Supplementary Appendices A3b, c), leading to the formation of bizarre crystal shapes (Supplementary Appendices A3g, h, i). Therefore, the new surface of the inner wall of the straw channel is extremely rugged with smooth crystal faces and deep depressions at their contacts (Supplementary Appendix A3g–i). Such an environment is sensitive to the sorption of impurities and to the attack of dissolved aggressive components (e.g., Stigliano et al. 2021). In addition to corrosion, blockage of the central canal by calcite growth (Fig. 2d) is equally devastating – stopping the seepage water flow. Such straws could stop growing or drop due to increased hydrostatic pressure. Growth of the individual calcite crystals in the straw channel requires special conditions: Different orientations of the crystals indicate independent nucleation of each crystal from highly supersaturated water coming from upper passages (at least episodically). This requires free spaces above the cave into which the seepage water degasses (see e.g., according to Eq. (1) (from right to left) (e.g., Pracný et al. 2016a).
Corrosion is generally initiated in places with excess surface energy (pores, kinks, edges of crystal layers, dislocations, and other defects). Etched pits appeared on the surface of the calcite standards at the sites of point defects (pores and various lattice defects) (Fig. 6a and b). Micromorphology of this corrosion is similar to the pitting corrosion known from metals (Ansari et al. 2018) or silicates / oxides (Lasaga and Blum 1986). Details on the morphology of etch pits on calcite can be found, e.g., in Macinnis and Brantley (1992) or Yoshino et al. (2010). The longitudinal grooves were formed at locations of line defects (edges of crystalline layers, dislocations) (Fig. 6a, white arrows). The advanced stage of corrosion has already progressed along well-developed crystal faces, which were less soluble than the surrounding material. This results in conservation of the inner structure of calcite/limestone with well-contoured rhomboscalenohedral (scalenorhombohedral) to scalenohedral calcite (Fig. 4a–d, or Fig. 5c–f). This was confirmed by the results of experimental corrosion of limestone (Fig. 7d–f) and calcite standards (Fig. 8d and e). The only difference is the better arrangement of scalenohedral crystals in calcite standards. In limestone, individual enclaves of scalenohedral calcites show a different orientation at larger distances (Fig. 7d and e).
The extent/level of corrosion may be viewed as a quantity of dissolved calcite during a time period. At derivation of the expression for the rate of calcite dissolution, we proceeded from the empirical rate law, R = k (1 − Ω)n, where R is the rate of dissolution in [mol m−2 s−1] units, k is rate constant in the same units, Ω is saturation state (see Eq. 8), and n is empirical coefficient. The rate R may be further broken down to R = − dm/(A dt), where m is the quantity of dissolved calcite [mol], A is the interaction area [m2], and t is the interaction time [s]. A combination of both expressions for R with consequent recalculation gives an expression in integral form for the calculation of dissolved calcite mass in time:
where t1–t0 is the time period of interaction. The saturation state Ω can be roughly constant under the open system conditions (fresh water still flowing in) or decrease with reaction progress under the conditions of the closed system (conditions similar to a batch rector). Similarly, area A could be assumed to be roughly constant for a short period of time. For a long period, area A would be dependent on t or m. It can be deduced that the corrosion rate could decrease with time due to the need to transport the dissolution products away from the solid surface.
A radical change in the corrosion mechanism occurs in extremely aggressive waters. Under these conditions, corrosion does not respect the crystal structure of the solids and proceeds en-bloc, as documented by the artificial dissolution of limestone standards in 0.1 mol L−1 HCl (Supplementary Appendix A6). Such corrosion would lead to smoothing of the speleothem surface rather than selective dissolution. It should be pointed out that such conditions are improbable under normal karst conditions. However, this smoothing is in some cases visible on some speleothems demonstrably in contact with unsaturated water.
Based on our results, a new model of corrosion associated with clay–calcite interaction was proposed as a competing reaction in which dissolved Ca2+ is exchanged for an alkali metal ion (Na+, K+) at the water–clay (ion exchanger) boundary:
The detailed mechanism is shown below: calcite dissolves according to Eq. (2) to the equilibrium given by Eq. (7). Parallel to this process, an ion exchange occurs, during which the Ca2+ ion binds to a clay mineral, e.g., Na-beidellite:
Binding of Ca upsets the balance in the solution described by Eqs. (2) and (7), and the loss of calcium is compensated by further dissolution (corrosion) of calcite. Smectite (alkaline beidellite or montmorillonite) is a suitable ion exchanger (e.g., Zeyen et al. 2022). The results of experimental corrosion of limestone standards in three types of clays under cave conditions are presented in Fig. 9. Different stages of corrosion are visible in dependence on the rate of the process: starting pitting corrosion (Fig. 9a, b, and f) up to advanced corrosion (Fig. 9c, d, and e). Examples of corrosion associated with speleothem clay contamination are given in Supplementary Appendix A5-3.
Corrosion mechanisms and conditions
The results of this work suggest that all types of corrosion can be included in one single mechanism: attack of the calcite/limestone surface by an aggressive unsaturated water/solution according to Eqs. 11, 12, 13. The intensity of corrosion primarily depends on the degree of aggressiveness of the water expressed by the saturation index (Eq. 10) of Ω (Eq. 8), with thermodynamics being the ultimate driving force of the process. Another important factor is the interaction time, see Eq. 15. The corrosion evolves in time from pitting corrosion to massive corrosion, affecting large areas on the surface and progressing deeper into the mineral/rock. The extent of corrosion is controlled mainly by the time of interaction and also by the local resistance of a given material in carbonate. It increases in the range: calcite cement > small calcite particles (micrite component) ≥ crystal faces with defects > surface of well crystallized calcite without defects (sparite enclaves, calcite monocrystals in soda straw wall). Whereas there is a single basic mechanism of corrosion (see Eqs. 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14), there are multiple mechanisms of unsaturated water formation. Generally, seven main categories of conditions supporting corrosion (CSC) may be distinguished based on previously published works and our results/experiences:
CSC-1: High dynamics of seepage water
Seepage waters can be divided into six categories based on their dynamics; see the classification of flow types from their discharge and discharge variability in Smart and Friederich (1987) and Baker et al. (1997). If the flow rate through the vadose zone exceeds the rate of processes driving water saturation with calcite/carbonates (Eqs. 5, 10, 11, and 12), the seepage water enters the cave unsaturated. The drip in the cave then shows a strongly variable discharge, dependent on rainfalls. Currently, this mechanism is studied in the Zemní pyramidy dome (Amatérská cave, MK). A highly variable discharge of 376 ± 241 ml h−1 (confidence interval at α = 0.05) and a low saturation index SI = − 0.21 ± 0.07 correlating with the specific electrical conductivity SEC = 403 ± 20 μS cm−1 (all based on a 30-month data set) are clearly consistent with the CSC-1 mechanism (Pracný et al. 2022).
CSC-2: insufficient water–limestone contact
Seepage water is isolated from the carbonate rocks along the flow path (e.g., by a layer of organic substances, a layer of clays, etc.). This hypothesis is an alternative to the CSC-1 hypothesis if the percolating/infiltrating water shows little dependence on variations in the surface conditions (precipitation). Currently, we have studied this mechanism in the chimney “Pršavý komín” (Býčí Skála cave, MK). This drip shows a relatively stable discharge with an average value of 3048 ± 135 ml h−1. Water is unsaturated with respect to calcite in the warmer half of the year (April to October) while slightly supersaturated in the colder (November to March), with the saturation index (SI(calcite)) ranging widely from − 0.52 to 0.68 with an average value of SI(calcite) = 0.04 ± 0.26 (all based on a data set covering one year). SI(calcite) is correlated with the specific electrical conductivity, SEC = 286 ± 12 μS cm−1 (Pracný et al. 2022). We believe that the water flow is isolated from the limestone by a layer of clay from a layer of the Rudice Beds stratigraphically above the cave. Microscopic images of naturally corroded limestone from the “Pršavý komín” are in Fig. 5. The samples show selective corrosion, in which some structural components of the limestone (micrite) are dissolved. In contrast, other components (sparite) are relatively resistant, leading to a pattern that follows the original limestone texture. Recrystallization is visible in some places on the surface (see Fig. 5e and f). Comparison with artificially corroded limestone standards (Fig. 7) shows considerable similarity, especially with respect to the conservation of scalenohedral calcite. Artificially corroded calcite (Fig. 8) shows better crystal arrangement (Fig. 8d and e), which can be expected due to perfect calcite crystallization.
CSC-3: mixing of different waters
An increase in water aggressivity by mixing two waters of different geochemical properties (Bögli 1964; Ernst 1964) has been known for a long time. It is often used in models of primary cave formation (Dreybrodt et al. 2010; Gabrovšek and Dreybrodt 2000, 2010, 2021). However, this mechanism can also be applied to two different types of water formed under different conditions and seeping vertically into the cave. Waters saturated under different \({\text{P}}_{\text{CO}_2}\) are a typical example: suppose that the first water infiltrated in a place without soil cover (\({\text{P}}_{\text{CO}_2}=10^{-3.5}\)) mixes with the second water infiltrated in a place with soil cover (\({\text{P}}_{\text{CO}_2}=10^{-1.5}\)). Although both initial solutions would be saturated with calcite, mixing increases the aggressivity of the mix (SIcalcite decreases) due to the nonlinearity of the equilibrium curve. The dependence of key parameters of the resulting mixture on the mixing ratio is shown in Appendix A7. This mixture achieves the highest aggressivity (SI(calcite) ~ − 0.55) at a mixing ratio of 9–1 (0.9 parts of water carbonated under \({\text{P}}_{\text{CO}_2} {\sim}10^{-3.5}\) and 0.1 parts of water carbonated under \({\text{P}}_{\text{CO}_2} {\sim}10^{-1.5}\)). A similar example can be mixing allochthonous water (water coming from a non-karst area) with vertically seeping water.
CSC-4: water condensation
Water condenses from water vapor on solid surfaces cooler than the dew point. An example of water condensation on various surfaces from the entrance passages of the Býčí skála cave is shown in Supplementary Appendix A8-1. The condensed water is saturated with CO2 at the \({\text{P}}_{\text{CO}_2}\) of the cave air but unsaturated with calcite. This water is strongly aggressive to carbonates, causing a process known as condensation corrosion (Tarhule-Lips and Ford 1998; Dublyansky and Spötl 2015; Liñán et al. 2021; White et al. 2021): see consequences of condensation corrosion in limestone (Supplementary Appendix A8-2). Condensation is controlled by the relative humidity of the air and the temperature difference between the air and the solid surface (speleothems, cave walls). The dew point determines the temperature at which water condenses. Suppose the cave air temperature is, for example, 8 °C and the relative humidity is 95%. The water will condense at a temperature of 7 °C (on the surface of a cooler limestone wall, floor, or a speleothem). The dependence of the dew point on temperature at constant humidity (calculated based on Lawrence 2005) is shown in Supplementary Appendix A9.
Allochthonous waters were found to play a significant role in condensation corrosion: Summer floods (see Supplementary Appendix A10) transport warm water into the cave, significantly increasing the temperature / humidity of the cave air, while the cave walls remain relatively cool and become condensation surfaces. In addition, if the flood flow becomes turbulent, the water degasses, increasing CO2 concentration in the air. This CO2 dissolves in condensed water and increases its aggressivity. A typical consequence of this process is the corroded limestone wall in front of the Velká Síň chamber (Býčí skála, MK) (Supplementary Appendix A11).
CSC-5: side reactions
CSC-5A: reactions binding calcium
The hypothesis of carbonate corrosion by clay was formulated on the basis of observations in caves (see Supplementary Appendix A5-3) before the corrosion was experimentally confirmed (Fig. 9). The proposed mechanism, consisting of binding of dissolved Ca2+ ions to smectite as an ion exchanger, is an original idea (Eqs. 17 and 18) and requires further study, e.g., simulation of the process in the laboratory under well-defined conditions. Many yet unknown relationships should be explored, such as the total dynamics of the process, the role of the exchange capacity of different clays, or the possibility of a ‘regeneration’ of clay exchangers under natural cave conditions.
CSC-5B: reactions lowering pH
These conditions are associated with metal oxidation in a muddy environment (see, Gázquez et al. 2013): iron (II) and manganese (II) are minor but abundant elements contained in clays and limestone that are released during dissolution. These ions are unstable under oxidizing conditions: they oxidize, thereby releasing H+ ions and acidifying the environment:
and
H+ ions increase the corrosion rate; see Eq. 11.
This mechanism was tested through experimental corrosion of limestone standards in muds marked as SS-Fe-clay (Býčí skála cave, MK). This clay was chosen based on the typical rusty color, which suggests a high iron content. However, the subsequent phase analysis did not confirm a higher content of usual Fe-minerals, see Table 2. Consequently, tested standards showed the lowest corrosion levels of all examined clays in the form of very slight pit corrosion (Fig. 9f).
CSC-6: effect of bacteria and organic matter
An example of cave microbiology is given, e.g., in Cunningham et al. (1995). In general, there is a consensus that organisms can influence rock weathering; see the review of Northup and Lavoie (2001) or Barton and Northup (2007). In the case of carbonates, bacteria excreting CO2 or/and organic acids could increase the dissolution rate through local changes in pH (see Eq. 11). However, some authors have referred to organisms rather as having an inhibitory effect on the formation of etch pits and, consequently, on dissolution rate (Lüttge and Conrad 2004). The inhibition effect is probably caused by the colonization of surfaces by bacteria with the development of monolayer biofilms on the substrate surface. Studies of surface interactions between microbes and carbonate minerals have traditionally focused on the precipitation of carbonate rather than the dissolution. Corrosion by bat guano is another specific mechanism of biogenic corrosion. We did not focus on this mechanism as its manifestations in the caves of the Moravian Karst are inconspicuous; interested parties are referred to the works of Dandurand et al. (2019), Audra et al. (2019), (2021) or Sala et al. (2023).
CSC-7: generally overrated and improbable mechanisms
CSC-7A: transfer of non-carbonate acidity from the surface to the cave
The transfer of non-carbonate acidity through the carbonate environment is unlikely, although this idea is fairly widespread among local cavers. There is no conclusive study that would demonstrate this mechanism. Preservation of acidity throughout the water flow from the surface would depend on the balance between the dynamics of water flow and the kinetics of neutralization reactions (according to Eq. 11). In principle, the residence time of water in the overburden would have to be shorter than the time required to neutralize the water acidity. Therefore, an extremely fast water flow, similar to the CSC-1 mechanism, or an isolating effect, as in the CSC-2 mechanism, would be necessary. The natural source of non-carbonate acidity in the vadose zone is mainly fulvic acids produced by biodegradation of organic matter (see, e.g., Qualls 2005; Ma et al. 2016). It is controlled by the type of vegetation with decreasing acidity in the sequence: coniferous > deciduous > grass. However, this acidity is already neutralized in the soil profile or the epikarst by interactions with limestone clasts (Ličbinská 2011). Moreover, according to sporadic publications, humic substances have a rather inhibitory effect on the surface of calcite (Compton and Sanders 1993). Another source of non-carbonate acidity (largely anthropogenic) can be hydrated oxides (SOX and NOX) in acid rain (Wu et al. 2012; Yu et al. 2015) or aggressive organic and inorganic fertilizers (Tkaczyk et al. 2020), with the same caveats as the natural sources. Some cavers in the Moravian Karst tried to associate the transfer of acidity with the falling of soda straws. However, this idea was refuted by an extensive study that explained the fall of soda straws by mechanical damage of the straw's inner wall (presence of abundant cracks in the straw calcite wall) (Faimon et al. 2004, 2006).
CSC-7B: increase in anthropogenic CO2 levels in caves
The concern that CO2 exhaled by cave visitors can transform (super)saturated drip water into unsaturated (aggressive) water is relatively widespread. However, this process is very unlikely. Water is formed in the epikarst at \({\text{P}}_{\text{CO}_2} {\sim}10^{-1.9}\) to 10−1.0 (Faimon et al. 2012a; Pracný et al. 2016b). \({\text{P}}_{\text{CO}_2}\) in cave air is usually much lower, reaching up to 10−2. This difference represents a driving force for water degassing that leads to water supersaturation and growth of calcite speleothems. Conversion of drip water to an unsaturated state would require the cave \({\text{P}}_{\text{CO}_2}\) to exceed \({\text{P}}_{\text{CO}_2}\) in the epikarst (\({\text{P}}_{\text{CO}_2}\) of water formation). However, such an increase associated with exhaled CO2 has not yet been documented. An analysis of this problem can be found in Lang et al. (2017). The situation is diametrically different in the case of condensed water: additional dissolution of CO2 under increased \({\text{P}}_{\text{CO}_2}\) (see Eqs. 1 and 12) significantly increases water aggressivity.
CSC-7C: other influences
Other compounds, such as nitrates, sulfates, chlorides, pesticides, metals, etc., are routinely monitored in seepage water (drip water). However, these components generally do not react with carbonates and cannot directly cause the corrosion of calcite speleothems. Strictly speaking, these compounds could shift chemical equilibria through changes in activity coefficients caused by increases in the ionic strength (see, e.g., Palmer et al. 1984) or through the complexation of solid surfaces (see, e.g., Ruiz-Agudo et al. 2009; Klasa et al. 2013; Heberling et al. 2021). However, this effect will probably be very low under usual karstic conditions. Therefore, these compounds are more important in the context of water quality than in the case of speleothems corrosion.
Recently, Kukuljan et al. (2021) published a model in which low-calcium water seeping through the vertical karst profile is on its flow path additionally saturated by high-level \({\text{P}}_{\text{CO}_2}\), transforming the water into undersaturation with respect to calcite. In such a way, a modified drip could dissolve the rock surfaces and speleothems on the cave floor. According to the authors, the model requires the thin rock overburden above the cave and a very specific airflow path. It does not seem to be a common mechanism in carbonate karst, where the highest \({\text{P}}_{\text{CO}_2}\) is usually observed in soil/epikarst. Percolating water saturated by carbonates at this soil/epikarst \({\text{P}}_{\text{CO}_2}\) on its further flow path rather degasses CO2 instead of an additional dissolution. Therefore, this mechanism was included among “Other influences”.
Some authors distinguish so-called mechanical corrosion (e.g., Wróblewski et al. 2017). It represents a mechanical abrasion of a solid surface by small to larger clasts transported by water flow. No doubt, this mechanism dominates especially during the primary development of caves. At present, it occurs during floods, which, however, cause more significant damage, such as contamination of speleothem with clays and their breaking by water pressure. We have already published a very specific case of corrosion/abrasion by blast furnace slag (Faimon et al. 2022a). Generally, there is a question of whether to call the mechanical damage “corrosion".
Conceptual model
Conditions supporting speleothem corrosion may be demonstrated on a conceptual model based on a typical karst vertical profile (Supplementary Appendix A12). Key components of the profile are (1) soils, (2) epikarst with a perched aquifer and a CO2 gas collector (forming an open system), (3) mixing zone of seepage water (vadose zone, forming a closed system) and (4) cave microclimate characterized by the variables such as CO2 level, temperature, humidity, and air flow (cave ventilation). All these variables control the growth/corrosion of calcite / speleothems. The CO2 gas collector is meant to be a layer of sediment or soil with increased CO2 concentrations in its free pores. Recently, a similar collector was proven by drilling on the Harbechy plateau (MK) at depths of around 3 m, lying at the base of epikarst, on the borderline of loess soils and underlying limestones (Faimon et al., unpublished data). This gas collector is roughly consistent with the underground CO2 of Atkinson (1977).
According to the specific processes and predominant types of corrosion, the vertical profile can be divided into four main zones. Zone A is classically considered to be the primary source of CO2 driving the karst processes, as shown by Eqs. 1, 2, 3, 4, 5 and 12. Soil CO2 is produced by biochemical decay of organic matter and respiration of the plant root system (e.g., Kuzyakov 2006; Faimon and Lang 2018). However, recent works indicate that the main sources of CO2 could lie deeper in the vadose zone (e.g., Benavente et al. 2010). This is consistent with our idea of a gas collector at the interface between the epikarst and limestone bedrock (zone B). This idea is conditioned by the flushing of organic matter from soils deeper into the subsoil and supported by tillage. In the zones A and B, karst water chemistry is formed according to Eqs. 1, 2, 3, 4, 5 and 11, 12, 13, 14. Zone C, the site of the mixing of waters flowing through the vadose zone by matrix pores and the crack system, can be the site of the further modification of water composition, the increase of its aggressiveness by mixing corrosion, both immediately in the vadose zone or/and later in the cave. The dynamic of water flow and the contact with limestone is crucial for corrosion. Zone D represents the microclimatic environment of the cave. The processes running here are (a) drip water degassing (Eq. 1 from right to left, in dependence on partial pressures \({\text{P}}_{\text{CO}_2}\) in water and cave air), (b) control of CO2 levels via the fluxes from the cave overburden and cave ventilation in dependence on ventilation modes (Faimon et al. 2012b), (c) changes in temperature and relative humidity controlling condensation process. The impact of visitors could be substantial.
Summary of anthropogenic impacts
Based on the corrosion-promoting conditions and mechanisms listed above, the potential anthropogenic influences were deduced as follows:
Seepage water flow rate exceeding the rate of water saturation (CSC-1) is mainly determined by the structure/morphology of the epikarst and vadose zone. Anthropogenic influences could be introduced via changes in infiltration conditions, e.g., through construction works in the epikarst or vadose zone. In the case of insufficient contact (CSC-2), the critical process is the transport of clays and fine sediments through the fracture system and their deposition along the transport path. Anthropogenic impact can be associated with the mobilization of clay material and with the support of its faster infiltration from the surface into the subsoil, e.g., agricultural activities such as filling and cultivating sinkholes or other field openings in the subsoil. Mixing of waters (CSC-3) can be significantly influenced by anthropogenic activity (see, e.g., Baker and Genty 1998). It is potentially devastating to cause formation of different types of infiltrated water due to a local loss of soil layer, for example, by cultivating unsuitable agricultural products (e.g., maize) or by deforestation. Water condensation (CSC-4) depends on the microclimatic conditions in the cave. It can be anthropogenically influenced in many ways: First, cave visitors participate in water condensation by exhaling water vapor. Second, humans influence cave ventilation by making cave corridors accessible, excavating additional cave entrances, and building modifications of cave spaces. Even moving visitors can change ventilation (Lang et al. 2015). Ventilation influences the variables controlling condensation, such as temperature, humidity, and the dew point. Corrosion in tourist caves associated with anthropogenic impact on cave microclimate is an example (Hoyos et al. 1998; Sánchez-Moral et al. 1999; Martín-García 2010). The side reactions (CSC-5) depend on natural conditions, especially the content and composition of clays and muds (Fe and Mn). It is difficult to imagine any anthropogenic influence, except perhaps mobilization of these materials and their transport into the cave, for example, by changes to water flow. The effect of bacteria associated with CSC-6 clearly requires more microbiological studies. The anthropogenic impact can easily be predicted: It would be associated with contamination caused by municipal / industrial wastewater and the excessive use of natural fertilizers in agriculture. Regarding CSC-7, the transfer of acidity from the surface to the cave is unlikely. However, some caution is needed with the selection, maintenance, and management of cultivated vegetation with respect to the CSC-3 mechanism. It is necessary to bear in mind that increasing soil acidity will always contribute to faster denudation of surface limestones. The increase in CO2 concentration caused by exhalation is dangerous primarily in synergy with water condensation: the aggressiveness of water increases with the concentration of dissolved CO2, which is controlled by PCO2 in cave air. The effect of additional foreign substances (both organic and inorganic), often of anthropogenic origin, is undesirable from the point of view of water purity and its potential use as drinking water; the effect of the compounds on corrosion is small or negligible. It should be remembered that the individual factors causing corrosion do not act in isolation, but that many of them overlap and are difficult to distinguish.
Conclusions
The study of corrosion of speleothems and carbonate rocks in epigenic caves of the Moravian Karst allowed us to draw the following conclusions:
-
The degree of corrosion is proportional to the product of water aggressiveness and interaction time. Local resistance of carbonate components with different crystallinities also plays a role.
-
The initial stages of corrosion are associated with pitting and longitudinal corrosion in the places of both point and line defects on a solid surface.
-
More advanced stages of corrosion are associated with corrosion of the entire surface, whereas the corroded material’s texture can be partially preserved.
-
Extremely aggressive water does not respect the internal structure of the corroded material and proceeds to depth en bloc.
-
Conditions favoring corrosion are: (1) high dynamics of seepage water, (2) insufficient water–limestone contact, (3) mixing of different waters, (4) water condensation, and (5) side reactions. Bacterial influence was not part of the study and requires additional research.
-
Concerns about the transfer of surface acidity into the cave, the increase in anthropogenic CO2 levels (except for their effect on condensation corrosion), and interactions of various foreign compounds in seepage water (mostly non-reactive with carbonates) seem to be exaggerated.
-
A new side reaction that binds dissolved calcium and increases the aggressivity of water was proposed and tested in a cave environment.
-
Possible anthropogenic impacts are: (1) construction works or agricultural interventions on the surface that change the geomorphology of subsoil, mobilizing clay and some other materials by faster infiltration, (2) activities allowing formation and infiltration of different types of water associated with changes in the vegetation cover, loss of soil profile, etc., (3) changes of cave microclimate via cave ventilation, e.g., by excavating additional entrances, removing sediment to make corridors passable, building modifications of cave spaces, exhalation of water vapor by visitors, and even visitor movement changing the air flow, (4) contamination caused by municipal / industrial wastewater and excessive use of natural fertilizers.
Data availability
Data will be made available upon request.
References
Ansari TQ, Xiao Z, Hu S et al (2018) Phase-field model of pitting corrosion kinetics in metallic materials. NPJ Comput Mater 4:38. https://doi.org/10.1038/s41524-018-0089-4
Appelo CAJ, Postma D (2005) Geochemistry, groundwater and pollution, 2nd Edition. pp. 638, CRC Press, London, https://doi.org/10.1201/9781439833544
Arvidson RS, Ertan IE, Amonette JE, Luttge A (2003) Variation in calcite dissolution rates: a fundamental problem? Geochim Cosmochim Acta 67:1623–1634. https://doi.org/10.1016/S0016-7037(02)01177-8
Atkinson TC (1977) Carbon dioxide in the atmosphere of the unsaturated zone: an important control of groundwater hardness in limestones. J Hydrol 35:111–123
Audra P, Palmer AN (2011) The pattern of caves: controls of epigenic speleogenesis. Geomorphologie: Relief. Process Environ 17:359–378. https://doi.org/10.4000/geomorphologie.9571
Audra P, D’antoni-Nobecourt J-C, Bigot J-B (2010) Hypogenic caves in France. Speleogenesis and morphology of the cave systems. Bull De La Société Géologique De France 184:327–335. https://doi.org/10.2113/gssgfbull.181.4.327
Audra P, De Waele J, Bentaleb I et al (2019) Guano-related phosphate-rich minerals in European caves. Int J Speleol 48:75–105. https://doi.org/10.5038/1827-806X.48.1.2252
Audra P, Heresanu V, Barriquand L et al (2021) Bat guano minerals and mineralization processes in Chameau Cave, Eastern Morocco. Int J Speleol 50:91–109. https://doi.org/10.5038/1827-806X.50.1.2374
Baker A, Genty D (1998) Environmental pressures on conserving cave speleothems: effects of changing surface land use and increased cave tourism. J Env Manag 53:165–175. https://doi.org/10.1006/jema.1998.0208
Baker A, Barnes WL, Smart PL (1997) Variations in the discharge and organic matter content of stalagmite drip waters in Lower Cave. John Wiley & Sons, Bristol
Barton HA, Northup DE (2007) Geomicrobiology in cave environments: past, current and future perspectives. J Cave Karst Stud 69:163–178
Benavente J, Vadillo I, Carrasco F et al (2010) Air carbon dioxide contents in the vadose zone of a mediterranean karst. Vadose Zone J 9:126–136. https://doi.org/10.2136/vzj2009.0027
Blecha M, Faimon J (2014) Spatial and temporal variations in carbon dioxide (CO2) concentrations in selected soils of the Moravian Karst (Czech Republic). Carbon Evapor 29:395–408. https://doi.org/10.1007/s13146-014-0220-7
Bögli A (1964) Mischungskorrosion – ein beitrag zum verkarstungsproblem. Erdkunde 18:83–92. https://doi.org/10.3112/erdkunde.1964.02.02
Busenberg E, Plummer LN (1986) A comparative study of the dissolution and crystal crowth kinetics of calcite and aragonite. In: Mumpton FA (ed) Studies in diagenesis, U.S. Geological Survey Bulletin. pp 139–168
Cailhol D, Audra P, Nehme C et al (2019) The contribution of condensation-corrosion in the morphological evolution of caves in semi-arid regions: preliminary investigations in the kyrenia range, cyprus. Acta Carsologica 48:5–27. https://doi.org/10.3986/ac.v48i1.6782
Carrasco ISS, Reis A, Fábio DA (2021) Modeling the kinetics of calcite dissolution in neutral and alkaline solutions. Geochim Cosmochim Acta 292:271–284. https://doi.org/10.1016/j.gca.2020.09.031
Chou L, Garrels RM, Wollast R (1989) Comparative study of the kinetics and mechanisms of dissolution of carbonate minerals. Chem Geol 78:269–282. https://doi.org/10.1016/0009-2541(89)90063-6
Columbu A, Audra P, Gázquez F et al (2021) Hypogenic speleogenesis, late stage epigenic overprinting and condensation-corrosion in a complex cave system in relation to landscape evolution (Toirano, Liguria, Italy). Geomorphology 376:107561. https://doi.org/10.1016/j.geomorph.2020.107561
Compton RG, Sanders GHW (1993) Dissolution of calcite in aqueous acid: the influence of humic species. J Colloid Interface Sci 158:439–445
Cunningham KI, Northup DE, Pollastro RM et al (1995) Bacteria, fungi and biokarst in Lechuguilla Cave, Carlsbad Caverns National Park, New Mexico. Env Geol 25:2–8. https://doi.org/10.1007/BF01061824
Dandurand G, Duranthon F, Jarry M et al (2019) Biogenic corrosion caused by bats in Drotsky’s Cave (the Gcwihaba Hills, NW Botswana). Geomorphology 327:284–296. https://doi.org/10.1016/j.geomorph.2018.10.027
Dreybrodt W (1981) Kinetics of the dissolution of calcite and its applications to karstification. Chem Geol 31:245–269
Dreybrodt W, Romanov D, Kaufmann G (2010) Evolution of caves in porous limestone by mixing corrosion: a model approach. Geologia Croatica 63:129–135. https://doi.org/10.4154/gc.2010.09
Dreybrodt W, Fohlmeister J (2022) The impact of outgassing of CO2 and prior calcium precipitation to the isotope composition of calcite precipitated on stalagmites. Implications for reconstructing climate information from proxies. Chem Geol 589:120676. https://doi.org/10.1016/j.chemgeo.2021.120676
Dublyansky YV, Spötl C (2015) Condensation-corrosion speleogenesis above a carbonate-saturated aquifer: Devils Hole Ridge, Nevada. Geomorphology 229:17–29. https://doi.org/10.1016/j.geomorph.2014.03.019
Ernst L (1964) Zur Frage Der Mischungskorrosion. Die Höhle 015:71–75
Faimon J, Lang M (2018) What actually controls the minute to hour changes in soil carbon dioxide concentrations? Geoderma 323:52–64. https://doi.org/10.1016/j.geoderma.2018.02.048
Faimon J, Zimák J, Zajíček P et al (2004) The study of recent destructive processes in the Moravian Karst caves (Czech Republic). Geographica (palacký University, Olomouc) 38:9–13
Faimon J, Ličbinská M, Zajíček P, Sracek O (2012) Partial pressures of CO2 in epikarstic zone deduced from hydrogeochemistry of permanent drips, the Moravian Karst, Czech Republic. Acta Carsologica 41:47–57
Faimon J, Troppová D, Baldík V, Novotný R (2012) Air circulation and its impact on microclimatic variables in the Císařská Cave (Moravian Karst, Czech Republic). Int J Climatol 32:599–623. https://doi.org/10.1002/joc.2298
Faimon J, Baldík V, Buriánek D et al (2022) Historical ferrous slag induces modern environmental problems in the Moravian Karst (Czech Republic). Sci Tot Env 847:157433. https://doi.org/10.1016/j.scitotenv.2022.157433
Faimon J, Baldík V, Kryštofová E, Štelcl J, Rez J (2022) Calcite raft formation in abandoned technical adit (Moravian Karst). Appl Geochem 141:105282
Faimon J, Štelcl J, Zimák J (2006) Recentní krasové procesy – destrukce speleotém (in Czech). Final Report, GAČR 205/03/1128. Brno
Frisia S (2015) Microstratigraphic logging of calcite fabrics in speleothems as tool for palaeoclimate studies. Int J Speleol 44:1–16. https://doi.org/10.5038/1827-806X.44.1.1
Frisia S, Borsato A, John Hellstrom J (2018) High spatial resolution investigation of nucleation, growth and early diagenesis in speleothems as exemplar for sedimentary carbonates. Earth-Sci Rev 178:68–91. https://doi.org/10.1016/j.earscirev.2018.01.014
Frumkin A, Aharon S, Davidovich U et al (2018) Old and recent processes in a warm and humid desert hypogene cave: ‘A’rak Na‘asane, Israel. Int J Speleol 47:307–321. https://doi.org/10.5038/1827-806X.47.3.2178
Gabrovšek F, Dreybrodt W (2000) Role of mixing corrosion in calcite-aggressive H2O-CO2-CaCO3 solutions in the early evolution of karst aquifers in limestone. Water Resour Res 36:1179–1188. https://doi.org/10.1029/1999WR900337
Gabrovšek F, Dreybrodt W (2010) Karstification in unconfined limestone aquifers by mixing of phreatic water with surface water from a local input: A model. J Hydrol 386:130–141. https://doi.org/10.1016/j.jhydrol.2010.03.015
Gabrovšek F, Dreybrodt W (2021) Early hypogenic carbonic acid speleogenesis in unconfined limestone aquifers by upwelling deep-seated waters with high CO2 concentration: A modelling approach. Hydrol Earth Syst Sci 25:2895–2913. https://doi.org/10.5194/hess-25-2895-2021
Gázquez F, Calaforra JM, Forti P et al (2013) Corrosion of calcite crystals by metal-rich mud in caves: Study case in Crovassa Ricchi in Argento Cave (SW Sardinia, Italy). Geomorphology 198:138–146. https://doi.org/10.1016/j.geomorph.2013.05.022
Genty D, Deflandre G (1998) Drip flow variations under a stalactite of the Père Noël cave (Belgium). Evidence of seasonal variations and air pressure constraints. J Env Manag 53:165–175. https://doi.org/10.1006/jema.1998.0208
Heberling F, Klačić T, Raiteri P et al (2021) Structure and Surface Complexation at the Calcite(104) – Water Interface. Environ Sci Technol 55:12403–12413. https://doi.org/10.1021/acs.est.1c03578
Hoyos M, Cañaveras JC, Sánchez-Moral S et al (1998) Microclimatic characterization of a karstic cave: human impact on microenvironmental parameters of a prehistoric rock art cave (Candamo Cave, northern Spain). Springer-Verlag
Kalová J, Mareš R (2015) Reference values of surface tension of water. Int J Thermophys 36:1396–1404. https://doi.org/10.1007/s10765-015-1907-2
Klasa J, Ruiz-Agudo E, Wang LJ et al (2013) An atomic force microscopy study of the dissolution of calcite in the presence of phosphate ions. Geochim Cosmochim Acta 117:115–128. https://doi.org/10.1016/j.gca.2013.03.025
Klimchouk A, Palmer AN, De Waele J et al (2017) Hypogene Karst Regions and Caves of the World. Springer, Tuscaloosa
Kottek M, Grieser J, Beck C et al (2006) World map of the Köppen-Geiger climate classification updated. Meteorol Z 15:259–263. https://doi.org/10.1127/0941-2948/2006/0130
Kukuljan L, Gabrovšek F, Johnston VE (2021) Low-calcium cave dripwaters in a high CO2 environment: Formation and development of corrosion cups in Postojna cave. Slovenia Water (switzerland) 13:3184. https://doi.org/10.3390/w13223184
Kuzyakov Y (2006) Sources of CO2 efflux from soil and review of partitioning methods. Soil Biol Biochem 38:425–448. https://doi.org/10.1016/j.soilbio.2005.08.020
Lang M, Faimon J, Ek C (2015) A case study of anthropogenic impact on the CO2 levels in low-volume profile of the Balcarka Cave (Moravian Karst, Czech Republic). Acta Carsologica 44:71–80
Lang M, Faimon J, Pracný P, Kejíková S (2017) A show cave management: Anthropogenic CO2 in atmosphere of Výpustek Cave (Moravian Karst, Czech Republic). J Nat Conserv 35:40–52. https://doi.org/10.1016/j.jnc.2016.11.007
Lasaga AC, Blum AE (1986) Surface chemistry, etch pits and mineral-water reactions. Geoch Cosmochim Acta 50:2363–2379
Laurent D, Durlet C, Barré G et al (2021) Epigenic vs. hypogenic speleogenesis governed by H2S/CO2 hydrothermal input and Quaternary icefield dynamics (NE French Pyrenees). Geomorphology 387:107769. https://doi.org/10.1016/j.geomorph.2021.107769
Laurent D, Durlet C, Barré G et al (2023) Unravelling biotic versus abiotic processes in the development of large sulfuric-acid karst. Geology 51:262–267. https://doi.org/10.1130/G50658.1
Lawrence MG (2005) The Relationship between relative humidity and the dewpoint temperature in moist air. A simple conversion and applications. Am Meteo Soc 86:225–233. https://doi.org/10.1175/BAMS-86-2-225
Ličbinská M (2011) Vegetation cover influence on geochemistry of recent karst processes. Faculty of Science, Masaryk University, Thesis
Liñán C, Benavente J, del Rosal Y et al (2021) Condensation water in heritage touristic caves: Isotopic and hydrochemical data and new approach for its quantification through image analysis. Hydrol Process 35:e14083. https://doi.org/10.1002/hyp.14083
Liss PS, Slater PG (1974) Flux of gasses across the air-sea interface. Nature 247:181–184. https://doi.org/10.1038/247181a0
Lüttge A, Conrad PG (2004) Direct observation of microbial inhibition of calcite dissolution. Appl Environ Microbiol 70:1627–1632. https://doi.org/10.1128/AEM.70.3.1627-1632.2004
Ma L, Xiao B, Di X et al (2016) Characteristics and distributions of humic acids in two soil profiles of the southwest China Karst area. Acta Geochimica 35:85–94. https://doi.org/10.1007/s11631-015-0086-y
Macinnis IN, Brantley SL (1992) The role of dislocations and surface morphology in calcite dissolution. Geochim Cosmochim Acta 56:1113–1126
Martín-García R, Martín-Pérez A, Alonso-Zarza AM (2011) Weathering of host rock and corrosion over speleothems in Castañar cave, Spain: an example of a complex meteoric environment. Carbon Evapor 26:83–94. https://doi.org/10.1007/s13146-010-0039-9
Martín-García R, Martín-Pérez A, Alonso-Zarza AM (2010) Petrological Study as a Tool to Evaluate the Degradation of Speleothems in Touristic Caves,Castañar de Ibor Cave, Cáceres, Spain. In: Andreo B, Carrasco F, Durán JJ, LaMoreaux JW (eds). Springer Berlin Heidelberg, Berlin, Heidelberg, p 509
Morse JW, Arvidson RS (2002) The dissolution kinetics of major sedimentary carbonate minerals. Earth Sci Rev 58:51–84
Musil R et al (1993) Moravský kras – labyrinty poznání. GEO program Adamov, Adamov
Northup DE, Lavoie KH (2001) Geomicrobiology of caves: a review. Geomicrobiol J 18:199–222. https://doi.org/10.1080/01490450152467750
Palmer AN (1991) Origin and morphology of limestone caves. Geol Soc Am Bull 103:1–21. https://doi.org/10.1130/0016-7606(1991)103%3c0001:OAMOLC%3e2.3.CO;2
Palmer DA, Ramette RW, Mesmer RE (1984) Triiodide ion formation equilibrium and activity coefficients in aqueous solution. J Solution Chem 13:637–683. https://doi.org/10.1007/BF00650374
Parkhurst DL, Appelo CAJ (2013) Description of input and examples for PHREEQC version 3. In A computer program for speciation, batch-Reaction, one-Dimensional transport, and inverse geochemical calculations. U.S. geological survey techniques and methods, book 6, chap. A43. Denver: US Department of the Interior, US Geological Survey
Peng C, Crawshaw JP, Maitland GC, Trusler JPM (2015) Kinetics of calcite dissolution in CO2-saturated water at temperatures between (323 and 373)K and pressures up to 13.8MPa. Chem Geol 403:74–85. https://doi.org/10.1016/j.chemgeo.2015.03.012
Plan L, Tschegg C, De Waele J, Spötl C (2012) Corrosion morphology and cave wall alteration in an Alpine sulfuric acid cave (Kraushöhle, Austria). Geomorphology 169–170:45–54. https://doi.org/10.1016/j.geomorph.2012.04.006
Plummer LN, Wigley TML, Parkhurst DL (1978) The kinetics of calcite dissolution in CO2 –water systems at 5 degrees to 60 degrees C and 0.0 to 1.0 atm CO2. Am J Sci 278:179–216. https://doi.org/10.2475/ajs.278.2.179
Pouchou JL, Pichoir F (1991) Quantitative analysis of homogeneous or stratifiedmicrovolumes applying the model “PAP.” In: Heinrich KFJ, Newbury DE (eds) Microprobe Quantitation. Plenum Press, pp 31–75
Pracný P, Faimon J, Sracek O, Kabelka L et al (2016) Anomalous drip in the Punkva caves (Moravian Karst): relevant implications for paleoclimatic proxies. Hydrol Process 30:1506–1520. https://doi.org/10.1002/hyp.10731
Pracný P, Faimon J, Kabelka L, Hebelka J (2016) Variations of carbon dioxide in the air and dripwaters of Punkva Caves (Moravian Karst, Czech Republic). Carbon Evapor 31:375–386. https://doi.org/10.1007/s13146-015-0259-0
Pracný P, Synková V, Roubal Z, Faimon J, Szabó Z, Kadlec R, Lang M (2022). Continuous monitoring of the hydrogeochemical properties of the Amatérská cave waters. 14th Scientific Conference “Research, Use and Protection of Caves”, Liptovský Mikuláš, September 6–8, 2022, Slovakia. In: Aragonit 27(1), 46–47 (Abstracts). ISSN 1335–213X. http://www.ssj.sk/user_files/Aragonit_27_1web.pdf
Qualls RG (2005) Biodegradability of fractions of dissolved organic carbon leached from decomposing leaf litter. Environ Sci Technol 39:1616–1622. https://doi.org/10.1021/es049090o
Rickard DT, Sjoberg EL (1983) Mixed kinetic control of calcite dissolution rates. Am J Sci 283:815–830. https://doi.org/10.2475/ajs.283.8.815
Rietveld HM (1969) A profile refinement method for nuclear and magnetic structures. J Appl Crystalography 2:65–71
Ruiz-Agudo E, Putnis CV, Jiménez-López C, Rodriguez-Navarro C (2009) An atomic force microscopy study of calcite dissolution in saline solutions: the role of magnesium ions. Geochim Cosmochim Acta 73:3201–3217. https://doi.org/10.1016/j.gca.2009.03.016
Sala P, Bella P, Postawa T et al (2023) Corrosion of carbonate speleothems by bat guáno. Sed Geol 454:106454. https://doi.org/10.1016/j.sedgeo.2023.106454
Sala P, Bella P (2023) Corrosion of carbonate speleothems by an allogenic river inferred from petrography and a weight loss experiment: a case study from the Demänová Cave System, Slovakia. Ann Soc Geol Polon https://doi.org/10.14241/asgp.2023.15
Sánchez-Moral S, Soler’ V, Canaveras JC, et al (1999) Inorganic deterioration affecting the Altamira Cave, N Spain: quantitative approach to wall-corrosion (solutional etching) processes induced by visitors. Sci Tot Env 243(244):67–84
Schott J, Brantley S, Crerar D et al (1989) Dissolution kinetics of strained calcite. Geochim Cosmochim Acta 53:373–382
Smart PL, Friederich H (1987) Water movement and storage in the unsaturated zone of a maturely karstified carbonate aquifer, Mendip Hills, England. In: Environmental problems in karst terranes and their solution. National Water Well Association, Bowling Green, Kentucky, pp 59–87
Stigliano L, Siena M, Ackerer P et al (2021) Statistical description of calcite surface roughness resulting from dissolution at close-to-equilibrium conditions. ACS Earth Space Chem 5:3115–3129. https://doi.org/10.1021/acsearthspacechem.1c00226
Stumm W, Morgan JJ (1996) Aquatic Chemistry: Chemical equilibria and rates in natural waters (3rd edition). 1040 pp. Wiley-Interscience Publication, John Wiley & Sons, Inc., New York, Chichster, Brisbane, Toronto, Singapore. ISBN-13: 978–0471511854
Subhas AV, Rollins NE, Berelson WM et al (2015) A novel determination of calcite dissolution kinetics in seawater. Geochim Cosmochim Acta 170:51–68. https://doi.org/10.1016/j.gca.2015.08.011
Tarhule-Lips RFA, Ford DC (1998) Condensation corrosion in caves on Cayman Brac and Isla de Mona. J Cave Karst Stud 60:84–95
Tkaczyk P, Mocek-Płóciniak A, Skowrońska M et al (2020) The mineral fertilizer-dependent chemical parameters of soil acidification under field conditions. Sustainability (switzerland) 12:7165. https://doi.org/10.3390/su12177165
Veetil SP, Mucci A, Arakaki T (2018) Dolomite dissolution kinetics in aqueous solutions in the presence of organic and inorganic additives at 25 °C and PCO2 ~ 1 atm. Chem Geol 483:98–110. https://doi.org/10.1016/j.chemgeo.2018.02.025
White JH, Domínguez-Villar D, Adam Hartland A (2021) Condensation corrosion alters the oxygen and carbon isotope ratios of speleothem and limestone surfaces. Results Geochem 2:100008. https://doi.org/10.1016/j.ringeo.2021.100008
Wróblewski W, Gradziński M, Motyka J, Stankovič J (2017) Recently growing subaqueous flowstones: Occurrence, petrography, and growth conditions. Quatern Int 437:84–97. https://doi.org/10.1016/j.quaint.2016.10.006
Wu Q, Han G, Tao F, Tang Y (2012) Chemical composition of rainwater in a karstic agricultural area, Southwest China: the impact of urbanization. Atmos Res 111:71–78. https://doi.org/10.1016/j.atmosres.2012.03.002
Xu J, Fan C, Teng HH (2012) Calcite dissolution kinetics in view of Gibbs free energy, dislocation density, and PCO2. Chem Geol 322–323:11–18. https://doi.org/10.1016/j.chemgeo.2012.04.019
Yoshino T, Kagi H, Kamiya N, Kokawa R (2010) Relation between etch-pit morphology and step retreat velocity on a calcite surface in aspartic acid solution. J Cryst Growth 312:1590–1598. https://doi.org/10.1016/j.jcrysgro.2010.01.041
Yu S, Kuo YM, Du W et al (2015) The hydrochemistry properties of precipitation in karst tourism city (Guilin), Southwest China. Environ Earth Sci 74:1061–1069. https://doi.org/10.1007/s12665-015-4235-8
Zeyen N, Wang B, Wilson SA et al (2022) Cation exchange in smectites as a new approach to mineral carbonation. Front Clim 4:913632. https://doi.org/10.3389/fclim.2022.913632
Acknowledgements
The authors are grateful to the Nature Conservation Agency of the Czech Republic and Cave Administration of the Czech Republic for wide support. Four anonymous reviewers are thanked for their comments that helped us substantially improve the manuscript.
Funding
Open access publishing supported by the National Technical Library in Prague. This research was funded by the Technology Agency of the Czech Republic, project Rock Environment and Natural Resources (RENS) No. SS02030023. The Czech Geological Survey, Masaryk University, and Brno University of Technology provided additional institutional support.
Author information
Authors and Affiliations
Contributions
Conceptualization: JF; supervision: JF; data curation: JF, VB, JŠ, DV, ZR, ZS, RH, PP; formal analysis: JF, PP, JR; funding acquisition: VB, RN, JF; investigation: JF, JŠ, VB; methodology: JŠ, JF, ZR, ZS; project administration: RN; resources: JF; validation: JF, PP, JR; visualization: JF, JR, RN; writing—original draft: JF; writing—review and editing: JF, PP, JR, ML, DV. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interests
The authors have no relevant financial or nonfinancial interests to disclose.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Faimon, J., Baldík, V., Štelcl, J. et al. Corrosion of calcite speleothems in epigenic caves of Moravian Karst (Czech Republic). Environ Earth Sci 83, 184 (2024). https://doi.org/10.1007/s12665-024-11449-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12665-024-11449-w