
Abstract
Elymus sensu stricto includes tetraploid species in the tribe Triticeae with a StH genome combination derived from Hordeum (H) and Pseudoroegneria (St). The group is related to many important cereals and forage grasses and is considered an important wild genetic resource for crop breeding. The relationships within Elymus s.s. have been difficult to resolve due to the large number of widely distributed species, a high degree of morphological diversity, introgression between species, and potential multiple origins. Using genome-wide DArTseqLD data from 57 taxa, including the StH group and associated species, it is possible to gain representative information about the genetic structure of the genus. SplitsTree, STRUCTURE and principal component analysis (PCoA) show that Elymus s.s. is divided into two major clades highly correlating with geographical origin separating American species from Eurasians. These findings, together with the contribution of different Pseudoroegneria and Hordeum species to the two clades, support the conclusion that Elymus s.s. has at least two independent origins. The Eurasian and American clades are further divided into three and two subclades, respectively. The results show that Elymus species have migrated multiple times from North to South America and between America and Eurasia. The new findings could be used to develop a new classification of Elymus s.s. with sections based on phylogenetic data.
Similar content being viewed by others


Introduction
Elymus L. (wheatgrasses) are perennial grasses in the tribe Triticeae (Poaceae) related to some of the most important agricultural cereal crops, such as wheat, barley, and rye. Several Elymus species are used as forage crops today, and the genus has an ethnobotanical record as food, medicine, and constructing material (Frawley et al. 2020). The morphologically and physiologically diverse genus makes it interesting as a wild genetic resource in plant breeding. A clear understanding of the intergeneric structure and relationships is required for efficient and precise utilization and conservation. Research on Elymus diversity and evolution has evolved together with the development of new techniques and have now entered the field of genomics.
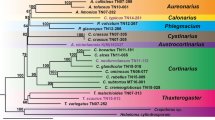
In most contemporary research, generic delimitations of Triticeae are based on genomic combinations where species with the same haplome or combination of haplomes are considered to belong to the same genus (Dewey 1984; Löve 1984; Baum et al. 2015). However, Elymus sensu lato (s.l.) has traditionally been an exception from this convention and includes allopolyploid species that originate from multiple intergeneric allopolyploidization events (Fig. 1). Elymus s.l. comprehends five different basic haplomes; St (Pseudoroegneria (Nevski) Á.Löve), H (Hordeum L.), P (Agropyron Gaertn.), W (Australopyrum (Tzvelev) Á.Löve), and Y (unknown origin) (Dewey 1984; Lu et al. 1993; Wang et al. 1994; Svitashev et al. 1996; Sun et al. 1997; Liu et al. 2006; Sun et al. 2008; Mason-Gamer et al. 2010a; Petersen et al. 2011). Genome symbols are based on Wang et al. (1994). The St-genome is the only common haplome represented in all Elymus s.l. species. The tetraploids have either a StH, StY or StP combination, while the hexaploid and octaploid species include StHH, StStHH, StStYY, StHY, StStY, StPY, StHW, and StWY (Lu 1993; Diaz 1999). The genome assignment to species is derived from cytological research but has been confirmed by genetic methods (Helfgott and Mason-Gamer 2004; Dong et al. 2015). The polyphyletic Elymus s.l. is often divided into a more consistent classification splitting the genome combinations into separate genera such as Roegneria K. Koch (StY), Kengyilia C. Yen & J. L. Yang (StPY), Campeiostachys Drobov (StHY), Anthosachne Steudel (StYW), Stenostachys Turcz. (StHW), and Elymus sensu stricto (s.s.) (StH) (Yen and Yang 2022). However, several groups are considered as sections rather than having generic status (Salomon and Lu 1992; Barkworth and von Bothmer 2009). The genus Pseudoroegneria includes 15–20 species, all distributed in central and eastern Asia except one, P. spicata, which is only found in North America (Dewey 1984; Carlson 1993). Genetic research has shown that Pseudoroegneria is the maternal St genome donor of both StStHH and StStYY (Mason-Gamer et al. 2002; McMillan and Sun 2004; Liu et al. 2006; Hodge et al. 2010; Dong et al. 2015). No Pseudoroegneria species occurs in South America and hence all Elymus species on the continent should originate from North America. Previous research found that gene transfer between North and South American Elymus is possible when grown together (Hunziker 1955, 1967; Dewey 1972, 1977; Jensen 1993).

Elymus s.s. (StH) is probably also a polyphyletic group with several involved progenitor species, but our understanding of polyploid evolution in the genus is still limited. Multiple origins have been suggested within single StH species. For example, Elymus caninus shows different versions of the RPB2 gene in both the St and the H genome (Yan and Sun 2012), and polyphyletic Hordeum-like sequences have been found in the Pepc tree of E. trachycaulus (Zuo et al. 2015). Earlier research also suggested multiple origins involving several Pseudoroegneria species (Liu et al. 2006, Sun et al. 2008, Fan et al. 2013b, Gao et al. 2015, Lei et al. 2020) and the differentiation of polyploid species with the St genome into three groups based on rDNA ITS sequences (Zhang et al. 2009).
Reticulate evolution through frequent interspecific hybridizations and introgressions is common within Elymus genome combinations altering species and population structures and contributing to the difficulties of solving the evolutionary history of the genus (Sun 2014; Baum et al. 2015; Wu et al. 2016). Moreover, these factors make it challenging to resolve the genetic relationships even within smaller species groups of Elymus, although attempts have been made (Helfgott and Mason-Gamer 2004, McMillan and Sun 2004, Mason-Gamer et al. 2005, 2010b, Liu et al. 2006, Sun et al. 2007, 2008, Mason-Gamer 2007, 2013, Petersen et al. 2011, Wang et al. 2012, Sun 2014, Baum et al. 2015, Gao et al. 2015). However, most research shows poorly resolved gene trees and incomplete sampling, which results in poorly resolved bifurcating trees.
Elymus s.s. (StH) occurs mainly in temperate areas in the northern and southern hemispheres from sea level up to 5 000 m in altitude (Dewey 1984; Löve 1984). The StH genome group contains approximately 50 species and is diverse in habitat preference and morphology (Sun and Salomon 2009). The genus is divided into mainly a boreal and temperate Eurasia-North American disjunction and a South American-North American disjunction (Thorne 1972). The relationships between these two disjunctions are not fully known. The main species diversity occurs in the northern hemisphere, but a high diversity can also be found in southern South America, especially in Chile and Argentina (Seberg and Petersen 1998, Leo et al. unpublished). Two species are circumpolar, E. alaskanus and E. macrourus, occurring in both North America and Eurasia. Also, E. sibiricus and E. trachycaulus are often considered circumpolar (Baum et al. 2016), though E. sibiricus in North America is probably recently introduced (Bennett 2006), and the main distribution of E. trachycaulus is in North America with only E. trachycaulus ssp. novae-angliae occurring in eastern Russia (Peschkova 1990).
Most research of the genetic relationships in Elymus include a small set of loci. This approach is limited by the risk of insufficient information and confusing results, especially in recently diverged lineages where incomplete lineage sorting and/or ongoing hybridization cause conflicting gene-trees (Choi et al. 2019). DArTseqLD™ is a high-throughput genotyping-by-sequencing platform using restriction enzymes to reduce genome complexity and sequencing of the restriction fragments to gain a large number of SNPs (Single Nuclear Polymorphisms) covering the whole genome (Jaccoud et al. 2001; Sansaloni et al. 2011). The technique has become popular for investigating diversity and phylogeny because of its cost efficiency, the high gain of informative and genome-wide molecular markers, and publicly available bioinformatics pipelines without the need of a reference genome.
The objectives of the present study were to investigate the genetic structure, geographic patterns of diversity, and the phylogeny within Elymus s.s. (StH). A clear genetic structure of the genus is important for the future establishment of a more robust circumscription and classification of the genus as well as a framework for further interspecific and population research. Additional objectives were to investigate the potential differentiation in parental origin within the StH combination and the relationships between the two disjunctions (North America-South America and America-Eurasia).
Material and methods
Plant material
This study included 282 individuals from 96 accessions (2–3 individuals per accession) representing 57 taxa from North America, South America, and Eurasia (Table 1). Most accessions are tetraploid Elymus s.s. (StH), but several associated species were included: 1) Three hexaploid Elymus s.s. (StHH) species and one accession of Elymus repens (StStH?) for origin analyses of hexaploids: 2. Six species with the StY genomes (referred to the genus Roegneria) for differentiation comparisons; 3. Three Pseudoroegneria species (St) and four Hordeum (H) species representing parental gnomes; 4. Three Eremium erianthum (N?) accessions, and one accession each of Psathyrostachys juncea (N) and Brachpodium sylvaticum from the tribe Brachypodieae used as more distantly related outgroups. The tetraploid StH Elymus accessions were selected to maximize the known taxonomical diversity and geographical distribution. The group constitutes several species with wide and narrow distribution ranges, but widespread species are more predominant in the current study. Seed samples were collected from the field or obtained from seed genebank material originally from wild native stands. The genebank material was sown in a greenhouse (20°C, no external light source) at the Swedish University of Agricultural Sciences (SLU). Leaf samples were taken at the seedling and young tillering stage from plants grown in the greenhouse and in the field, respectively, and immediately dried in silica gel (ca 1–3 mm aluminosilicate, Merck KGaA, Darmstadt, Germany). The samples were kept in silica gel until DNA extraction and genotyping. Plants grown in the greenhouse were kept until maturity for taxonomical verification.
Genotyping
The leaf samples were sent to Diversity Arrays Technology (DArT) Pty Ltd., Bruce, Australia, for DNA extraction and genotyping using DArTseqLD™ technology (Jaccoud et al. 2001; Sansaloni et al. 2011). The received data was filtered with the pipeline ‘dartR’ (Gruber et al. 2018) in R v. 4.1.1 (R core team 2021). The filtering setting affected the number of accessions in the analyses resulting in a loss of taxa. Therefore, two sets were created and referred to as Set 1 filtered to include all samples, and Set 2 including only tetraploid StH Elymus species. Both sets were filtered on: 1) reproducibility (a measure of the consistency of scoring technical replicates) with a threshold of ≥ 95%; 2) locus call rate with a threshold set to ≥ 50%; 3) random removal of all but one of the multiple SNPs in the same fragment to avoid the potential influence of linkage (only for the STRUCTURE analyses); 4) individual call rate with the threshold set to zero and ≥ 40% for Set 1 and Set 2, respectively; 5) removal of monomorphic loci; and 6) removal of loci with a minor allele frequency (MAF) ≤ 2%. The gl2svdquartets function in ‘dartR’ was used to generate NEXUS files for the genetic structure and relationship analyses, with SNPs converted to haploid states and heterozygous loci coded using standard ambiguity codes. Based on the results from Set 1 and 2, two additional filtering (Set 3 and 4) were conducted with the same setting as Set 2 representing the two major clades in Elymus s.s.. The raw read data is available through the NCBI Short Read Archive (SRA) under BioProject ID PRJNA942607.
PCoA and STRUCTURE analyses
To investigate the genetic structure of the genome combinations a Principal Component Analysis (PCoA) based on Nei´s unbiased genetic distance was carried out using the ade4 package in R (Dray et al. 2007), and visualized by the ggplot2 package (Villanueva and Chen 2019). The two data sets were analyzed using the software STRUCTURE v.2.3.4 (Pritchard et al. 2000) for genomic structure and to infer the number of the most likely groups within the StH genome combination. The method uses a Bayesian iterative algorithm to analyze differences in the distribution of genetic variants amongst populations by placing samples into groups whose members share similar patterns of variation. Each analysis was performed with 100 000 repetitions of the Markov Chain Monte Carlo (MCMC) with a burn-in of 50 000 interactions in 10 independent simulations, and without prior information to define the clusters. The number of clusters (K) was determined using the average likelihood values of the delta K method (Evanno et al. 2005) implemented in the program Structure Harvester (Earl and von Holdt 2012). Bar plots of the ancestry coefficients of each sample were created using ‘ggplot2’ in R (Wickham 2016; Villanueva and Chen 2019).
SplitsTree and coalescence-based (SVDquartets) analyses
To explore the complex relationships of Elymus s.s. and detect patterns of reticulation caused by hybridization and incomplete lineage sorting, an unrooted phylogenetic network analysis, including associated species, was performed for Set 1 and Set 2 using SplitsTree 4.0 (Huson and Bryant 2006). Default settings were used, implementing Neighbor-Net analysis with the variance of ordinary least squares, EqualAngle, Uncorrected P, and missing data treated as unknown. Bootstrapping was conducted with 10 000 replicates. Finally, two phylogenetic multispecies coalescent-based analyses with SVDquartets (Chifman and Kubatko 2014) in PAUP* v.4.0a169 (Swofford 2003) were performed to assess the relationships within the identified groups. Elymus sibiricus and E. confuses were used as an outgroup in the analysis of Eurasian group, and E. lanceolatus as an outgroup in the analysis of the American group. All quartets were evaluated, and branch support was assessed through 10 000 nonparametric bootstrap replicates. SVDquartets is computationally efficient when analyzing a large amount of SNP data under the MSC and can be applied without any loss of power to infer the true species tree (Wascher and Kubatko 2021).
Results
DArTseqLD™
The processed sequence dataset provided by DArT Pty Ltd. contained in total 282 genotypes with 20 790 codominant binary SNP markers and 48% missing data, which could be expected from next generation sequences analyses with a taxonomical diverse set of genotypes. For Set 1, the filtering removed 235 unreproducible loci (≤ 95%), 8 624 loci below 50% call rate, and 5017 minor alleles (≤ 2%), resulting in 6914 binary SNPs with 33% of missing data and all genotypes remained. Additionally, 3201 secondary loci were removed prior to the STRUCTURE analyses resulting in 3713 loci left in the analysis. Following initial exploratory analyses with SplitsTree and PCoA, one accession (CHL18-39 E. glaucescens) was identified as a hybrid accession and removed from subsequent analyses. The filtering in Set 2, using a threshold for individual call rate at ≥ 40%, resulted in 187 remaining genotypes with in total 6305 binary SNPs and 27% missing data. After the removal of 147 secondaries only, 4888 unlinked loci were left for the STRUCTURE analysis. In Set 3, including species in the Eurasian group, filtering resulted in 104 genotypes and 3591 SNP markers (22% missing), and Set 4, including species in the American group, 83 genotypes and 5589 SNP markers (25% missing data) remained. It was assumed that the amount of missing data could be tolerated without losing accuracy due to the large number of informative genetic markers in the analyses (Philippe et al. 2004).
Phylogenetic analyses
Set 1 and Set 2 were used for network analysis in SplitsTree (Figs. 2 and 3). The results from Set 1 showed a separation between StY and StH species with a further division of the StH species into two major groups, also evident in the analysis of Set 2. The clustering of the StH accessions is highly correlated with geographical origin. One group included all the American species, except E. kamczadalorum, not separating between North and South America. The other group included the Eurasian species, as well as the circumpolar species E. macrourus and E. alaskanus. Based on the assumption that the two groups found in the SplitsTree, PCoA and STRUCTURE analyses have independent origins, the SVDquartets analyses in PAUP* (Fig. 4) were performed independently for species in the American and Eurasian clade, respectively. The outgroups were chosen based on the SplitsTree phylogenetic network using Set 2.

SplitsTree phylogenetic network (Huson and Bryant 2006) reconstructed using Set 2, including tetraploid genotypes of StH Elymus species (n = 187) and 6 305 binary SNPs. Blue species names indicate South American origin

SplitsTree phylogenetic network (Huson and Bryant 2006) reconstructed using Set 1, including all genotypes, both StH and associated species (n = 282) and 6 914 binary SNPs

Phylogenetic trees based on SVDquartets, conducted with PAUP, using DArTseqLD™ SNP data with 10 000 nonparametric bootstrap replicates. The analysis was performed with species identified belonging to the Eurasian (Set 3) and American (Set 4) clade, respectively, with outgroup chosen based on the SplitsTree phylogenetic network using Set 2. Bootstrap values are indicated above branches and branches with a posterior probability < 0.75 are shown as polytomies. Blue species names indicate South American origin
In the SplitsTree analysis of Set 1, Pseudoroegneria spicata and the American Hordeum spp. were grouped together with the “American” Elymus StH species, while P. strigosa and H. roshevitzii grouped together with the Eurasian Elymus StH species. In contrast, P. ferganensis was intermediate and did not show clear affiliation to any group. The SplitsTree network placed Eremium erianthum together with Psathyrostachys juncea. Elymus repens appeared as an intermediate with a (partial) reticulate relationship between StY and StH genome combinations. The hexaploid E. scabriglumis appeared in a network associated with the American Hordeum group and the second American StH subgroup most closely related to E. trachycaulus.
The results from the Set 2 SplitsTree analysis and the SVD-quartet analyses showed that the “Eurasian” clade was further divided into three subgroups with high branch support (100% BS). The correlation between geography and the division into subgroups was not as distinct as for the two main groups. The first subgroup (from central and eastern Asia) included E. sibiricus with E. confusus as a sister group. The second subgroup (from primarily western and central Eurasia) included E. caninus, E. fibrosus, E. alopex, and E. charkeviczii clustering within most of the E. mutabilis accessions. The third subgroup included E. jacutensis, E. komarovii, E. sajanensis, E. transbaicalensis (from primarily eastern and central Asia), together with E. alaskanus E. macrourus (circumpolar species). The phylogenetic results could not separate between E. sajanensis and E. transbaicalensis. In the SplitsTree analysis, E. dentatus appeared as an intermediate between the second and third subgroups, but in the phylogenetic analysis the taxa were placed in the third subgroup. One individual of E. mutabilis (H10456) appeared as an intermediate between the “American” and “Eurasian” groups in the SplitsTree analysis of Set 2.
The “American” group was further divided into two main subgroups. The first subgroup included predominantly North American species including E. bakeri, E. kamczadalorum, E. trachycaulus, E. virescens, and E. violaceus. There was a clear relationship between Asian and American E. trachycaulus accessions except E. trachycaulus (H10514) forming a monophyletic group (100% BS) together with E. kamczadalorum from Kamchatka and E. virescens from Northern North America. The second subgroup included both South and North American species intermixed with weak internal branch support (< 70% BS) indicating unresolved polytomic relationships. However, accessions of E. angulatus formed a well-supported (100% BS) clade together with the two North American species E. villosus and E. glaucus. In addition, accessions of E. glaucescens and E. parodii formed a subclade (100% BS).
Analyses of genetic structure
The genetic structures in the PCoA and STRUCTURE analyses (Figs. 5 and 6) were consistent with the results from the SplitsTree and SVDquartets analyses. For Set 1, the first (PoC1 24%) and second (PCo2 12%) axis of variation showed an evident separation of the American StH, Eurasian StH and StY taxa. The PCoA analysis of Set 4 (PCo1 18%, PCo2 10%), including only American StH species, clearly separated the two subgroups as seen in the SplitsTree analyses. In addition, a third subgroup containing E. glaucescens and E. parodii was separated from the other species. The PCoA analysis of Set 3 (PCo1 28%, PCo2 20%), including only Eurasian StH species, showed a clear distinction between the three subgroups as seen in the SplitsTree analyses. The STRUCTURE analyses showed the existence of six (K = 6) and two groups (K = 2) for Set 1 and Set 2, respectively (Fig. 6), based on the ΔK. Samples with an ancestry coefficients > 0.75 and < 0.75 were assigned to as “pure” and “admixed”, respectively. Set 2 showed low or no admixture levels between the two StH groups. Based on both taxonomical grouping and the geographic distribution of the species, a substructure of the six STRUCTURE groups could be observed: K1 = StY, K2 = StH 2nd Eurasian subgroup, K3 = StH 1st American subgroup, K4 = StH 1st and 3rd Eurasian subgroup, K5 = Ns and Brachypodium, and K6 = StH 2nd American subgroup. Elymus repens showed a complete admixture. Elymus bakeri, E. violaceus, E. virginicus, E. canadensis and E. lanceolatus showed a partial introgression between the two American subgroups when analyzing Set 1 but not Set 2. For Set 2, the two groups were consistent with the geographical differentiation between the two major StH groups, and in agreement with the results from the SplitsTree and PCoA analyses. Only one individual of E. mutabilis (H10456) was placed as an admixed between the “American” and “Eurasian” groups in the STRUCTURE analysis of Set 2. The assignments of the hexaploid species confirmed the earlier findings from the SplitsTree analyses that E. scabriglumis is most closely related to species in the 1st American subgroup, E. patagonicus the 2nd American subgroup and E. transhyrcanus to the 3rd Eurasian subgroup.

Principal components analysis (PCoA) based on Nei´s unbiased genetic distance showing the dispersion of samples across the first two principal components of Set 1 (all samples)

STRUCTURE bar plots of the estimated membership coefficient (Q) for each individual for K = 6 and K = 2 based on STRUCTURE results for Set 1 (left) with all species included and Set 2 (right) with only StH species
Discussion
Based on a molecular approach using DArTseqLD™, this paper presents a genetic structure and phylogenetic analysis of the complex genus Elymus s.s and some associated species. The utilization of a large number of molecular markers, widely distributed over the genome and a large number of taxa from the whole distribution range vouches for an adequate representation of the generic diversity.
The distinct separation of StH and StY genome combinations has previously been demonstrated using molecular markers (Svitashev et al. 1996; Sun et al. 1997) and is also evident in the current study. These results further confirm the affiliation of included species to previously determined genome combinations. The SplitsTree networks together with the PCoA and STRUCTURE analyses in the present study differentiate between two major groups of Elymus s.s. which highly correlate with geographical distribution, the “American” group and the “Eurasian” group. The placement of different Pseudoroegneria and Hordeum species in the different groups suggests the involvement of several progenitor species, making Elymus s.s. a polyphyletic taxon with the potential differentiation in a St1H1 and a St2H2 group. These results are in agreement with earlier research, thereby suggesting that American and Eurasian StH species have evolutionary distinct origins (Jaaska 1992; Linde-Laursen et al. 1994; Dubcovsky et al. 1997; Sun et al. 2008; Sun and Ma 2009; Wang et al. 2012, Fan et al. 2013b). Hence, it contradicts previous results suggesting a common origin (Mason-Gamer et al. 2010b, Wang et al. 2011; Dong et al. 2015). Baum et al. (2015) suggest, based on research of variation in 5S nrDNA, that Elymus StH has multiple origins also within continents, but the results from the present study cannot confirm this. Only three species of Pseudoroegneria and four from Hordeum were included in this study, hence, the results are incomprehensive for the exact progenitor species determination, and other progenitor species cannot be excluded. Different cultivars of the H genome could however indicate introgression by subsequent backcrossing (Zuo et al. 2015), and reciprocal hybridization is possible to have occurred. The origin of the parental species correlates with geography and the origin of the included Elymus s.s. species. In addition, the SplitsTree network place the South American Eremium erianthum together with Psathyrostachys juncea, which supports the classification of Eremium erianthum as distinct from Elymus (Seberg and Linde-Laursen 1996; Svitashev et al. 1996). One individual of E. mutabilis in accession H10456 was interpreted as an intermediate between the “American” and “Eurasian” group in both the STRUCTURE and SplitsTree analysis. This accession derives from Kamchatka, eastern Russia, and the individual is interpreted as a hybrid between the two major StH groups. In all analyses, the other individuals in the accession group are placed together with the rest of E. mutabilis in the Eurasian subgroup. This proves the potential of hybridization between the St1H1 and St2H2 group but the degree of introgression is not evident.
The Eurasian tetraploid Elymus StH group
The first (sibiricus-confusus) and the second (mutabilis-fibrosus-caninus-alopex) subgroups in Eurasia contain well-separated species. Although distinct in morphology and ecology, the close connection between E. mutabilis, E. fibrosus, and E. caninus is evident in both the biosystematic study by Agafonov and Salomon (2002) and the current study. A high frequency of hybrids has been found in areas where E. caninus, E. fibrosus, and/or E. mutabilis grow together (Diaz 1999; Sun et al. 2001; Wu et al. 2016), which additionally indicates a high degree of introgression and a close relationship, although the species are genetically separate. The present study confirms the separation between E. mutabilis and E. transbaicalensis (Agafonov 2004; Leo et al. 2022), as well as the inclusion of E. charkeviczii in E. mutabilis (Agafonov et al. 2005). The species E. alopex, endemic to Iceland, was described by Salomon (2005), who proposed that it constitutes a deviating form of E. caninus. The results from the SplitsTree analyses suggest an intermediate relationship with E. mutabilis and E. caninus forming a network. Elymus alopex could be of hybrid origin, and several other described taxa are assumed to be E. caninus × E. mutabilis hybrids (Agafonov and Salomon 2002).
In contrast, the third (alaskanus-macrourus-transbaicalensis) subgroup in the Eurasian clade, including two circumpolar species, is diverse and taxonomically more challenging to handle. The close relationship between E. macrourus, E. komarovii, E. transbaicalensis, E. sajanensis, and E. alaskanus, is in agreement with morphological and isozyme data, as well as biosystematic research showing high crossability between the species (Agafonov et al. 1998; Agafonov 2004). Agafonov and Salomon (2002) conducted a biosystematic study with inter- and intraspecific crosses of Siberian Elymus StH taxa to establish a model of recombination gene pools (RGP) and introgression gene pools (IGP). They concluded that Russian populations of E. transbaicalensis, E. komarovii, and E. alaskanus are connected within the same RGP and in the same IGP as E. macrourus. The microevolutionary levels of differentiation between E. komarovii, E. transbaicalensis, E. kronokensis, and E. sajanensis have been studied by Agafonov et al. (2019). The species were morphologically variable, with many different biotypes showing complex genetic relationships. The phylogenetic analyses in the present study confirm the relationships and differentiate between E. alaskanus, E. macrourus, E. jacutensis, E. komarovii, with E. transbaicalensis and E. sajanensis in a polytomic clade suggesting incomplete lineage sorting and/or extensive hybridization.
The fact that the two true circumpolar species group in the same subclade is phylogeographically interesting. The transcontinental occurrence of circumpolar species shows that the Bering Sea is a possible dispersal route. No signs of introgression could, however, be found between circumpolar species and co-occurring species in North America. Elymus alaskanus is usually morphologically homogenous within regions and populations but variable over its whole distribution area (Wu et al. 2016). The results, however, show all included accessions forming a monophyletic clade with high branch support (100% BS).
The American tetraploid Elymus StH group
The first subgroup in the American clade gather many of the species often referred to as the E. trachycaulus complex (Barkworth 1994). Elymus kamczadalorum, E. bakeri, and E. virescens have all been treated as subspecies to E. trachycaulus (Tzvelev 1973; Löve 1984; Sun et al. 2006a). Sun and Ma (2009) concluded, based on nucleotide variation in chloroplast Asp(GUC)–Thr(GGU) intergenic region, that E. violaceus is genetically distinct. Seen in the STRUCTURE analyses, both E. violaceus and E. bakeri, have a certain influence of genetic structure from the second American subgroup indicating introgression which could explain the deviating variation from E. trachycaulus. The E. trachycaulus complex remains a taxonomical challenge despite many attempts to unravel the genetic relationships (Gupta et al. 1988; Gaudett et al. 2005; Sun and Li 2005; Sun et al. 2006a, b; Stevens et al. 2007; Sun 2007; Zuo et al. 2015). The present study, however, clearly excludes taxa such as E. alaskanus, E. macrourus, E. scribneri, and E. sierrae, which are sometimes considered conspecific (Barkworth 1994). The close relationship between E. kamczadalorum from Kamchatka and the North American species indicates a North American origin with a migration of the group over the Bering Sea. Further, the close relationship between Asian and American E. trachycaulus indicates a second migration event in the group. However, E. trachycaulus has been grown in eastern Eurasia for feeding purposes and the origin of the included population is uncertain.
In the second American subgroup, a polytomic pattern in the SplitsTree and phylogenetic analyses indicates incomplete linage sorting and potential introgression due to recent and radial speciation events. The unresolved branches make the origin and migration of the South American Elymus species uncertain. In all analyses, the two species E. villosus and E. glaucus, from eastern respectively western North America, group together with E. angulatus from southern South America in a well-supported monophyletic subclade. Thus, the four South American species in the American clade, E. angulatus, E. cordilleranus, E. glaucescens, and E. parodii constitute a non-monophyletic group indicating either multiple migration events from North to South or a single migration event with a re-colonization to North America by E. villosus and E. glaucus. In southern Chile, E. angulatus and E. glaucescens sometimes grow sympatric. Individuals from accession CHL18-39 were determined as E. glaucescens in the field, with single spikelets per rachis node. In the first exploratory analyses, the individuals in the accession appeared as hybrids between E. angulatus and E. glaucescens and were subsequently removed from further analyses. Introgression among South American Elymus species may obscure the true origin and relationships. Gene introgression through recurrent hybridization could promote a rapid adaptation of these species to different ecological habitats resulting in the formation of many endemic species or genotypes/ecotypes (Sun et al. 2008). The close relationship between E. virginicus and E. canadensis is confirmed by frequent hybrids occurring in areas where the species grow in sympatry, and introgression is assumed (Pohl 1959; Nelson 1978). As seen in the STRUCTURE analysis, a certain influence from the 1st American subgroup is also evident in E. virginicus and E. canadensis indicating introgression showing the evolutionary dynamics among North American Elymus species.
Origin of hexaploid species
The repeated formation of hexaploids is evident from the placement of the three included hexaploid species (E. transhyrcanus, E. scabriglumis, and E. patagonicus) in different phylogenetic groups. Elymus transhyrcanus group together with E. caninus in the Eurasian clade, while the other two included hexaploid species are found in the American clade. Elymus scabriglumis is placed within the first American subgroup in all analyses, apart from the other South American species, showing a close relationship with the Mexican E. vaillantianus and the North American E. trachycaulus. This suggests a potential origin of E. scabriglumis from those North American species and an independent migration from North to South America either before or after the hexaploidization event. Dewey (1977) found that E. tilcarensis (syn. E. scabriglumis (Seberg and Petersen 1998)) is more closely related to E. trachycaulus than E. lanceolatus, which further supports the conclusion of origin. The analyses here also show that the tetraploid E. angulatus is most closely related to the sympatrically grown hexaploid E. patagonicus, and hence E. angulatus is the most likely progenitor to E. patagonicus. The second genome donor is still unknown. Several potential Hordeum progenitor species occur in the same area as E. angulatus and E. patagonicus, and artificial hybrids between E. angulatus and some Hordeum species are rather easily obtained (Jacobsen and von Bothmer 1981; Seberg and von Bothmer 1991). Research on morphology, cytology, and reproductive compatibility (biosystematics) between E. scabriglumis and E. patagonicus show a genetic barrier between the two hexaploid species with sterile artificial hybrids, which further suggests separate origins and the involvement of different Elymus and/or Hordeum progenitors (Hunziker 1953; Nicora 1978).
Taxonomy
Phylogenetic and diversity data is essential for understanding relationships and evolution, but the results also have taxonomical implications. Petersen et al. (2011) argue that a classification of Elymus should be based on a combination of morphology and cytology as well as molecular data. The phylogenetic results from the present study provide new evidence for a revised classification with the implication of a division into subgenera (St1H1 and St2H2) and/or sections based on genetic relationships. The results recognize Dewey’s (1984) and Löve’s (1984) haplome delimiting criteria of Triticeae as an appropriate and pragmatic base for classification and the more consistent genera delimitation based on genome combination proposed by Yen and Yang (2022) as further criteria. However, the results recognize the phylogenetically inaccuracy in the classification system with the pronounced distinction between the two independent American and Eurasian Elymus s.s. groups that have originated from different progenitor species. The two major groups could be considered as subgenera, but it is then necessary to find morphological characteristics to distinguish them. The study cannot exclude several different origins also within the St1H1 and St2H2 groups.
In a diversity study including 24 StH species, Baum et al. (2016) used canonical discriminant analysis based on sequences of the nr5SDNA to identify factors related to genetic relationships and Elymus s.s. classification. They found evidence for a classification based on ploidy, section affiliation proposed by Yen and Yang (2022) (Elymus, Elytrigia, Goulardia, Hystrix, and Sitanion), and geographical regions (Eurasia, circumboreal, North America, and South America). The present study supports the geographical component on a large scale but does not differentiate between North and South America and does not support circumboreal species as a unified group even though the two true circumpolar species belong to the same phylogenetic subgroup. Polyploid species are not associated and group in different clades demonstrated by E. patagonicus, E. scabriglumis, and E. transhyrcanus with different parental origin. The sections earlier proposed by Yen and Yang (2022), Löve (1984), and Tzvelev (1976) are based on cytological and morphological characters (Tab. 1). Neither of the classifications are, however, in accordance with the phylogenetic results. For example, E. caninus (from Eurasia) and E. trachycaulus (from North America) are placed in the same section, Goulardia, by Löve (1984) and Yen and Yang (2022), but the two taxa fall into different clades in current phylogenetic analyses. These findings show the importance of integrative taxonomy, including taxonomical information from different data types and methodologies.
Conclusion
This study uses molecular DArTseqLD™ data to reveal the genetic structure and relationships within Elymus s.s. (StH) and some associated taxa. The analyses demonstrate a major division between predominantly American and Eurasian species with different parental species involved. Species from South America do not form a monophyletic group, which indicates multiple dispersal events from North to South America. The study manages to describe the major features of Elymus s.s. diversity but shows the difficulties in resolving evolutionary relationships even when using high throughput techniques with plenty of genetic markers. The complexity of Elymus s.s. relationships is shown in the poorly resolved internal branches, indicating a high degree of introgression, incomplete lineage sorting, recent speciation and multiple speciation events. In addition, phenotypic plasticity, cryptic speciation, few reliable morphological diagnostic characters, and a large number of taxa makes it taxonomically difficult to handle in practice. The technical advancements make access of genotypes the limiting factor rather than details of molecular markers. A larger number of species and populations could help resolving the polytomic relationships within the major clades and give a more elaborate understanding of the diversification process and the interspecific interactions among worldwide Elymus. In both species and section circumscriptions, molecular data should be used to reexamine morphological characters considered to be of diagnostic importance. A new classification of Elymus sections, where phylogenetic relationships are taken into account, is needed. The results from this study have given a deeper understanding of Elymus evolution and will form the basis for establishing a more accurate circumscription and classification of the genus and a robust framework for future interspecific and population studies.
Data availability
The datasets analysed during the current study are available in the NCBI Short Read Archive (SRA) repository under BioProject ID PRJNA942607.
References
Agafonov AV (2004) Intraspecific structure and reproductive relationships between Elymus mutabilis and E. transbaicalensis (Poaceae) in southern Siberia from the viewpoint of taxonomical genetics. Russ J Genet 40:1229–1238
Agafonov AV et al (2005) A biosystematic study of Elymus charkeviczii, E. mutabilis and E. subfibrosus from the Peninsula Kamchatka. Czech J Genet Plant Breed 41:79–85
Agafonov AV, Salomon B (2002) Genepools among SH genome Elymus species in boreal Eurasia. - In: Hernándes, P. et. al. (ed), Triticeae IV. Consejeria de Agricultura y Pesca. pp. 37–41.
Agafonov AV et al. (1998) Biosystematic relationships between Elymus komarovii (Nevski) Tzvel. and related species (AA Jaradat, Ed.). - Science Publishers Inc.
Agafonov AV et al (2019) The variability and specificity of the histone H1 among the Siberian species Elymus transbaicalensis, E. komarovii, E. sajanensis, E. kronokensis (Poaceae) and some morphologically deviating forms. Turczaninowia 22:5–18
Barkworth ME, von Bothmer R (2009) Scientific names in the Triticeae (C Feuillet and GJ Muehlbauer, Eds.). - Springer.
Barkworth ME (1994) The Elymus trachycaulus complex in North America: more questions than answers. - In: Wang, R. R. C. et al. (eds), Preceedings of the 2nd International Triticeae Symposium. Logan Press, pp. 189–198.
Baum BR et al (2015) Diversity within the genus Elymus (Poaceae: Triticeae) as investigated by the analysis of the nr5S rDNA variation in species with St and H haplomes. Mol Genet Genomics 290:329–342
Baum BR et al (2016) Diversity within the genus Elymus (Poaceae: Triticeae) II: analyses of variation within 5S nrDNA restrict membership in the genus to species with StH genomes. Mol Genet Genomics 291:217–225
Bennett BA (2006) Siberian wild rye (Elymus sibiricus L., Poaceae) in western North America: native or introduced? - Bot. Electron. News. #366.
Carlson J (1993) Pseudoroegneria (Nevski) Á. Löve. - In: Flora of North America Editorial Committee (ed), Flora of North America North and Mexico, Volume 24. in press.
Chifman J, Kubatko L (2014) Quartet Inference from SNP Data Under the Coalescent Model. Bioinformatics 30:3317–3324
Choi B et al (2019) Identifying genetic markers for a range of phylogenetic utility - From species to family level. PLoS One 14:e0218995–e0218995
Dewey DR (1972) Genome analysis of South American Elymus patagonicus and its hybrids with two North American and two Asian Agropyron species. Bot Gaz 133:436–443
Dewey DR (1977) Genome relations of South American Agropyron tilcarense with North American and Asian Agropyron species. Bot Gaz 138:369–375
Dewey DR (1984) The genomic system of classification as a guide to intergeneric hybridization with the perennial Triticeae. - Gene Manip. plant Improv.: 209–279.
Diaz O (1999) Genetic diversity in Elymus species (Triticeae) with emphasis on the Nordic region (Doctoral thesis). - Dep. Plant Breed. Res. Svalöv
Dong ZZ et al (2015) Phylogeny and differentiation of the St genome in Elymus L. sensu lato (Triticeae; Poaceae) based on one nuclear DNA and two chloroplast genes. BMC Plant Biol 15:179
Dray S et al (2007) The ade4 package-II: two-table and K-table methods. R news 7:47–52
Dubcovsky J et al (1997) Genome analysis of South American Elymus (Triticeae) and Leymus (Triticeae) species based on variation in repeated nucleotide sequences. Genome 40:505–520
Earl DA, von Holdt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the evanno method. Conserv Genet Resour 4:359–361
Evanno G et al (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Fan X et al (2013) Phylogenetic reconstruction and diversification of the Triticeae (Poaceae) based on single-copy nuclear Acc1 and Pgk1 gene data. Biochem Syst Ecol 50:346–360
Fan X et al (2013) Phylogeny and molecular evolution of the Acc1 gene within the StH genome species in Triticeae (Poaceae). Gene 529:57–64
Frawley ES et al (2020) An ethnobotanical study of the genus Elymus. Econ Bot 74:159–177
Gao G et al (2015) Phylogenetic relationships among Elymus and related diploid genera (Triticeae: Poaceae) based on nuclear rDNA ITS sequences. Biologia 70:183–189
Gaudett M et al (2005) Molecular variation and population structure in Elymus trachycaulus and comparison with its morphologically similar E. alaskanus. Plant Syst Evol 250:81–91
Gruber B et al (2018) Dartr: an r package to facilitate analysis of SNP data generated from reduced representation genome sequencing. Mol Ecol Resour 18:691–699
Gupta PK et al (1988) A stuy on genomic relationships of Agropyron trachycaulum with Elymus scabriglumis, Elymus innovatus and Hordeum procerum. Genome 30:525–528
Helfgott DM, Mason-Gamer RJ (2004) The evolution of North American Elymus (Triticeae, Poaceae) Allotetraploids: evidence from phosphoenolpyruvate carboxylase gene sequences. Syst Botany 29:850–861
Hodge CD et al (2010) Phylogenetic analysis of the maternal genome of tetraploid StStYY Elymus (Triticeae: Poaceae) species and the monogenomic Triticeae based on rps16 sequence data. Plant Sci 178:463–468
Hunziker JH (1953) Estudio citogenetico de un hibrido entre Elymus patagonicus y Agropyron agroelymoides. Rev Invest Agr 7:73–79
Hunziker JH (1955) Artificial and natural hybrids in the gramineae, tribe Hordeae, hybrids of Elymus and Agropyron. Am J Bot 42:459–467
Hunziker JH (1967) Chromosome and protein differentation in the Agropyron scabriglume complex. Taxon 16:259–266
Huson DH, Bryant D (2006) Application of phylogenetic networks in evolutionary studies. Mol Biol Evol 23:254–267
Jaaska V (1992) Isoenzyme variation in the grass genus Elymus (Poaceae). Hereditas 117:11–22
Jaccoud D et al (2001) Diversity Arrays: a solid state technology for sequence information independent genotyping. Nucleic Acids Res. 29:e25–e25
Jacobsen N, von Bothmer R (1981) Interspecific hybridization in the genus Hordeum L. - In: Whitehouse, R. N. H. (ed), Barley genetics IV. Proceeding. Edinburgh University Press, pp. 710–715.
Jensen KB (1993) Cytogenetics of Elymus magellanicus and its intrageneric and intergeneric hybrids with Pseudoroegneria spicata, Hordeum violaceum, Elymus trachycaulus, E. lanceolatus and E. glaucus (Poaceae, Triticeae). Genome 36:72–76
Lei YX et al (2020) Phylogenetic relationships and the maternal donor of Roegneria (Triticeae: Poaceae) based on three nuclear DNA sequences (ITS, Acc1, and Pgk1) and one chloroplast region (trnL-F). J Syst Evol 60:305–318
Leo J et al (2022) Population structure and phylogeography of Elymus mutabilis and its genetic relationships with E. transbaicalensis (Poaceae). Nord J Bot 2022:e03520
Leo J (2022) Phylogenetic studies of Elymus s.s. L. (Poaceae) : plant systematics as a prerequisite for efficient conservation and utilization. - Acta Univ. Agric. Sueciae in press.
Linde-Laursen IB et al (1994) Comparison of the Giemsa C-banded and N-banded karyotypes of two Elymus species, E. dentatus and E. glaucescens (Poaceae, Triticeae). Plant Syst Evol 192:165–176
Liu Q et al (2006) Phylogenetic relationships in Elymus (Poaceae: Triticeae) based on the nuclear ribosomal internal transcribed spacer and chloroplast trnL-F sequences. New Phytol 170:411–420
Löve A (1984) Conspectus of the Triticeae. Feddes Repert 95:425–521
Lu BR et al (1993) Biosystematic study of hexaploids Elymus tschimganicu and E. glaucisssimus. II. interspecific hybridization and genomic relationship. Genome 36:1157–1168
Lu BR (1993) Biosystematic investigations of asiatic wheatgrasses - Elymus L. (Triticeae: Poaceae) (Doctoral thesis). - Dep. Breed. Res. Svalöv
Mason-Gamer RJ (2007) Allopolyploids of the genus Elymus (Triticeae, Poaceae): a Phylogenetic Perspectiv. Aliso A J Syst Florist Bot 23:372–379
Mason-Gamer RJ (2013) Phylogeny of a genomically diverse group of Elymus (Poaceae) allopolyploids reveals multiple levels of reticulation. PLoS One 8:e78449
Mason-Gamer RJ et al (2002) Phylogenetic analysis of North American Elymus and the monogenomic Triticeae (Poaceae) using three chloroplast DNA data sets. Genome 45:991–1002
Mason-Gamer RJ et al (2005) Polyploidy, introgression, and complex phylogenetic patterns within Elymus. Czech J Genet Plant Breed 41:21–26
Mason-Gamer RJ et al (2010) Phylogenetic relationships and reticulation among Asian Elymus (Poaceae) allotetraploids: analyses of three nuclear gene trees. Mol Phylogenet Evol 54:10–22
Mason-Gamer RJ et al (2010) Reticulate evolutionary history of a complex group of grasses: phylogeny of Elymus StStHH allotetraploids based on three nuclear genes. PLoS One 5:e10989
McMillan E, Sun G (2004) Genetic relationships of tetraploid Elymus species and their genomic donor species inferred from polymerase chain reaction-restriction length polymorphism analysis of chloroplast gene regions. Theor Appl Genet 108:535–542
Nelson EN (1978) Hybridization and Introgression between Elymus canadensis and Elymus virginicus (Poaceae). Proc Oklahoma Acad Sci 58:32–34
Nicora EG (1978) Flora Patagonica. Parte III. Gramineae (M. N Correa, Ed.). - Instituto Nacional de Tecnologfa Agropecuaria.
Peschkova GA (1990) Elymus, L. - In: Malyschev, L. I. and Peschkova, G. A. (eds), Flora of Siberia, vol. 2, Poaceae (Gramineae). Scientific Publishers, Inc., pp. 361.
Petersen G et al (2011) The origin of the H, St, W, and Y genomes in allotetraploid species of Elymus L. and Stenostachys Turcz. (Poaceae: Triticeae). Plant Syst Evol 291:197–210
Philippe H, Snell EA, Bapteste E, Lopez P, Holland PW, Casane D (2004) Phylogenomics of eukaryotes: impact of missing data on large alignments. Mol Biol Evolut 21(9):1740–1752
Pohl RW (1959) Morphology and cytology of some hybrids between Elymus canadensis and E. virginicus. Proc Iowa Acad Sci 66:155–159
Pritchard JK et al (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Salomon B (2005) Deviating variants of Elymus caninus (Poaceae) in NW Europe. Willdenowia 35:245–251
Salomon B, Lu BR (1992) Genomic groups, morphology and sectional delimitaion in Eurasian Elymus (Poaceae, Triticeae). Plant Syst Evol 180:1–13
Sansaloni C et al (2011) Diversity arrays technology (DArT) and next-generation sequencing combined: genome-wide, high throughput, highly informative genotyping for molecular breeding of Eucalyptus. BMC Proc 5:P54
Seberg O, Linde-Laursen IB (1996) Eremium, a new genus of the Triticeae (Poaceae) from Argentina. Syst Bot 21:3–15
Seberg O, Petersen G (1998) A taxonomic revision of the genus Elymus L. s. l. (Poaceae, Triticeae) in South America. Bot Jahrbücher Fur Syst Pflanzengeschichte Und Pflanzengeographie 120:503–543
Seberg O, von Bothmer R (1991) Genome analysis of Elymus angulatus and E. patagonicus (Poaceae) and their hybrids with North American and South American Hordeum spp. Plant Syst Evol 174:75–82
Stevens L et al (2007) Microsatellite variability and heterozygote excess in Elymus trachycaulus populations from British Columbia in Canada. Biochem Syst Ecol 35:725–736
Sun GL (2007) Genetic diversity of rbcL gene in Elymus trachycaulus complex and their phylogenetic relationships to several Triticeae species. Genet Resour Crop Evol 54:1737–1746
Sun G (2014) Molecular phylogeny revealed complex evolutionary process in Elymus species. J Syst Evol 52:706–711
Sun G, Li W (2005) Molecular diversity of species of the Elymus trachycaulus species complex and their relationships to non-North American taxa. Plant Syst Evol 256:179–191
Sun GL, Ma XZ (2009) Nucleotide diversity and minisatellite in chloroplast Asp(GUC)-Thr(GGU) region in Elymus trachycaulus complex, Elymus alaskanus and Elymus caninus. Biochem Syst Ecol 37:67–75
Sun G, Salomon B (2009) Molecular evolution and origin of tetraploid Elymus species. Breed Sci 59:487–491
Sun GL et al (1997) Analysis of tetraploid Elymus species using wheat microsatellite markers and RAPD markers. Genome 40:806–814
Sun GL et al (2001) Genetic diversity and structure in a natural Elymus caninus population from Denmark based on microsatellite and isozyme analyses. Plant Syst Evol 227:235–244
Sun GL et al (2006) Molecular diversity and relationships among Elymus trachycaulus, E. subsecundus, E. virescens, E. violaceus, and E. hyperarcticus (Poaceae : Triticeae) as determined by amplified fragment length polymorphism. Genome 49:1160–1169
Sun G et al (2006) Molecular diversity and relationships of North American Elymus trachycaulus and the Eurasian E. caninus species. Genetica 127:55–64
Sun GL et al (2007) Molecular evolution and genome divergence at RPB2 gene of the St and H genome in Elymus species. Plant Mol Biol 64:645–655
Sun G et al (2008) Molecular phylogeny of RPB2 gene reveals multiple origin, geographic differentiation of H genome, and the relationship of the Y genome to other genomes in Elymus species. Mol Phylogenet Evol 46:897–907
Svitashev S et al (1996) A study of 28 Elymus species using repetitive DNA sequences. Genome 39:1093–1101
Swofford, D. L. 2003. PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods). in press.
Thorne RF (1972) Major disjunctions in the geographic ranges of seed plants. Q Rev Biol 47:365–411
Tzvelev NN (1973) A review of the tribe Triticeae Dum. from the family poaceae of the flora of the soviet union. Nov Sist Sosud Rast 10:19–59
Tzvelev NN (1976) Tribe 3: Triticeae Dum. - In: Poaceae U.R.S.S. Nacka Publishing House, pp. 146–298.
Villanueva RAM, Chen ZJ (2019) ggplot2: Elegant graphics for data analysis (2nd ed.). - Meas. Interdiscip. Res. Perspect. 17: 160–167.
Wang X-L et al (2011) Phylogeny of the StH genome species in Triticeae (Poaceae): evidence from chloroplast trnL-F and mitochondria COXII intron sequences. Biochem Syst Ecol 39:758–765
Wang X-L et al (2012) Phylogeny and molecular evolution of the DMC1 gene within the StH genome species in Triticeae (Poaceae). Genes Genom 34:237–244
Wang RC. et al. (1994) Genome symbols in Triticeae. - In: Wang, R. R. C. et al. (eds), Proceedings of the 2nd International Triticeae Symposium. Utha state University Press, pp. 29–34.
Wascher M, Kubatko L (2021) Consistency of SVDQuartets and maximum likelihood for coalescent-based species tree estimation. Syst Biol 70:33–48
Wickham H (2016) ggplot2: Elegant graphics for data analysis. - Springer-Verlag New York.
Wu PP et al (2016) Comparison of gene flow among species that occur within the same geographic locations versus gene flow among populations within species reveals introgression among several Elymus species. J Syst Evol 54:152–161
Yan C, Sun GL (2012) Multiple origins of allopolyploid wheatgrass Elymus caninus revealed by RPB2, PepC and TrnD/T genes. Mol Phylogenet Evol 64:441–451
Yen C, Yang JL (2009) Historical review and prospect of taxonomy of tribe Triticeae Dumortier (Poaceae). Breed Sci 59:513–518
Yen C et al (2005) Hitoshi Kihara, Askell Dive and the modern genetic concept of the genera in the tribe Triticeae (Poaceae). Acta Phytotaxon Sin 43:82–93
Yen C, Yang J (2022) Biosystematics of Triticeae, Volume II. Genera: Secale, Tritiosecale, Pseudosecale, Eremopyrum, Henrardia, Taeniantherum, Heteranthelium, Crithopsis, and Hordeum. - China Agriculture Press and Springer Nature Singapore Pte Ltd.
Zhang C et al (2009) Phylogenetic relationships among the species of Elymus sensu lato in Triticeae (Poaceae) based on nuclear rDNA ITS sequences. Russ J Genet 45:696–706
Zuo HW et al (2015) Origin and Reticulate Evolutionary Process of Wheatgrass Elymus trachycaulus (Triticeae: Poaceae). PLoS One 10:1–17
Funding
Open access funding provided by Swedish University of Agricultural Sciences. This work was supported by Kungliga Fysiografiska Sällskapet (Grant numbers 39809).
Author information
Authors and Affiliations
Contributions
All authors contributed to the study’s conception and design. Material preparation, data collection and analysis were performed by JL, TB, AM, ASC RvB. The first draft of the manuscript was written by JL and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
This work was supported by Kungliga Fysiografiska Sällskapet (Grant numbers 39809). The authors have no competing interests to declare that are relevant to the content of this article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Leo, J., Bengtsson, T., Morales, A. et al. Genetic structure analyses reveal multiple origins of Elymus sensu stricto (Poaceae). Genet Resour Crop Evol (2024). https://doi.org/10.1007/s10722-024-01944-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10722-024-01944-1