Journal of Computer-Aided Molecular Design ( IF 3.5 ) Pub Date : 2023-06-29 , DOI: 10.1007/s10822-023-00517-1 Gyoung Jin Park 1 , Nam Sook Kang 1
|
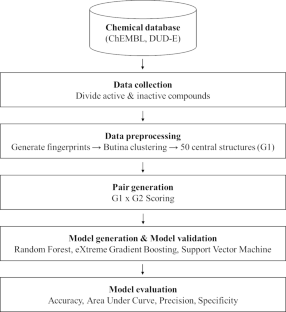
Drug candidates identified by the pharmaceutical industry typically have unique structural characteristics to ensure they interact strongly and specifically with their biological targets. Identifying these characteristics is a key challenge for developing new drugs, and quantitative structure-activity relationship (QSAR) analysis has generally been used to perform this task. QSAR models with good predictive power improve the cost and time efficiencies invested in compound development. Generating these good models depends on how well differences between “active” and “inactive” compound groups can be conveyed to the model to be learned. Efforts to solve this difference issue have been made, including generating a “molecular descriptor” that compressively expresses the structural characteristics of compounds. From the same perspective, we succeeded in developing the Activity Differences-Quantitative Structure-Activity Relationship (ADis-QSAR) model by generating molecular descriptors that more explicitly convey features of the group through a pair system that performs direct connections between active and inactive groups. We used popular machine learning algorithms, such as Support Vector Machine, Random Forest, XGBoost and Multi-Layer Perceptron for model learning and evaluated the model using scores such as accuracy, area under curve, precision and specificity. The results showed that the Support Vector Machine performed better than the others. Notably, the ADis-QSAR model showed significant improvements in meaningful scores such as precision and specificity compared to the baseline model, even in datasets with dissimilar chemical spaces. This model reduces the risk of selecting false positive compounds, improving the efficiency of drug development.
中文翻译:
ADis-QSAR:基于化合物生物活性差异的机器学习模型
制药行业确定的候选药物通常具有独特的结构特征,以确保它们与其生物靶标发生强烈且特异性的相互作用。识别这些特征是开发新药的关键挑战,定量构效关系(QSAR)分析通常用于执行这项任务。具有良好预测能力的 QSAR 模型可提高化合物开发投资的成本和时间效率。生成这些好的模型取决于“活性”和“非活性”化合物组之间的差异能够在多大程度上传递给要学习的模型。人们已经做出了努力来解决这个差异问题,包括生成压缩表达化合物结构特征的“分子描述符”。从同样的角度来看,我们成功地开发了活性差异-定量结构-活性关系(ADis-QSAR)模型,通过生成分子描述符,通过在活性基团和非活性基团之间执行直接连接的配对系统,更明确地传达基团的特征。我们使用支持向量机、随机森林、XGBoost 和多层感知器等流行的机器学习算法进行模型学习,并使用准确度、曲线下面积、精度和特异性等评分来评估模型。结果表明支持向量机的性能优于其他机器。值得注意的是,即使在化学空间不同的数据集中,与基线模型相比,ADis-QSAR 模型在精确度和特异性等有意义的分数方面也显示出显着的改进。该模型降低了选择假阳性化合物的风险,提高了药物开发的效率。


























 京公网安备 11010802027423号
京公网安备 11010802027423号