Adsorption ( IF 3.3 ) Pub Date : 2023-10-24 , DOI: 10.1007/s10450-023-00418-7 Shotaro Hiraide , Kohei Yamamoto , Hideki Tanaka , Kazuyuki Nakai , Satoshi Watanabe , Minoru T. Miyahara
|
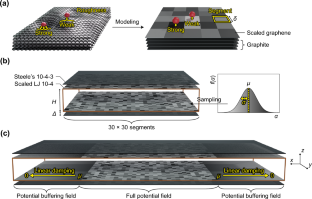
For porous carbons, which typically have hierarchical structures, the pore size distribution (PSD) is one of the most important characteristics and is currently evaluated by using kernel fitting methods represented by non-local density functional theory. Herein, we present new kernels for N\(_{2}\) and Ar adsorption at 77 K and 87 K, respectively, derived from Monte Carlo (MC) simulations based on a carbon slit-pore model that considers energetic heterogeneity due to surface roughness. The model consists of a locally scaled Lennard–Jones (LJ) 10-4 potential and Steele’s 10-4-3 potential, and the scaling factors of the LJ 10-4 potential are assumed to follow a normal distribution that mimics the adsorption behavior on real carbon black. In contrast to our previous MC kernel based on Steele’s 10-4-3 potential, the local isotherms of the new kernel did not show a steep increase due to adsorption layer formation. Despite the improved fit for adsorption isotherms, PSDs obtained from the proposed kernel unfortunately show a non-negligible valley around 1 nm, which is a major artifact of the kernel fitting approach. A careful comparison of the smooth and rough surface models indicated that the definitive cause of the artifact lies not in the formation of monolayers, which was believed so far, but rather in the pore-filling behavior, which provides a major clue for constructing a completely artifact-free kernel based on molecular simulations.
Graphical abstract
中文翻译:
基于考虑表面能量异质性的简化狭缝孔模型分析多孔碳孔径分布的 GCMC 核
对于通常具有分级结构的多孔碳,孔径分布(PSD)是最重要的特征之一,目前通过使用以非局域密度泛函理论为代表的核拟合方法来评估。在此,我们分别提出了 77 K 和 87 K 下 N \(_{2}\)和 Ar 吸附的新内核,这些内核是根据基于碳狭缝孔模型的蒙特卡罗 (MC) 模拟得出的,该模型考虑了由于以下原因引起的能量异质性:表面粗糙度。该模型由局部缩放的 Lennard–Jones (LJ) 10-4 势和 Steele 10-4-3 势组成,并且假设 LJ 10-4 势的缩放因子遵循正态分布,模拟了吸附行为真正的炭黑。与我们之前基于 Steele 10-4-3 势的 MC 内核相比,新内核的局部等温线并未因吸附层的形成而出现急剧增加。尽管吸附等温线的拟合得到了改进,但不幸的是,从所提出的核中获得的 PSD 显示了 1 nm 左右的不可忽略的谷,这是核拟合方法的主要伪影。对光滑和粗糙表面模型的仔细比较表明,伪影的最终原因不在于单分子层的形成,而单分子层的形成却被认为如此,而在于孔隙填充行为,这为构建完全的纳米结构提供了重要线索。基于分子模拟的无伪影内核。


























 京公网安备 11010802027423号
京公网安备 11010802027423号