Neurogenetics ( IF 2.2 ) Pub Date : 2023-10-26 , DOI: 10.1007/s10048-023-00737-5 Shintaro Aoki 1, 2 , Kazuki Watanabe 1 , Mitsuhiro Kato 3 , Yukihiko Konishi 4 , Kazuo Kubota 5, 6 , Emiko Kobayashi 7 , Mitsuko Nakashima 1 , Hirotomo Saitsu 1
|
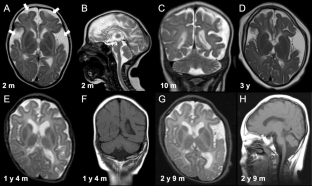
Sphingomyelin phosphodiesterase 4 (SMPD4) encodes a member of the Mg2+-dependent, neutral sphingomyelinase family that catalyzes the hydrolysis of the phosphodiester bond of sphingomyelin to form phosphorylcholine and ceramide. Recent studies have revealed that biallelic loss-of-function variants of SMPD4 cause syndromic neurodevelopmental disorders characterized by microcephaly, congenital arthrogryposis, and structural brain anomalies. In this study, three novel loss-of-function SMPD4 variants were identified using exome sequencing (ES) in two independent patients with developmental delays, microcephaly, seizures, and brain structural abnormalities. Patient 1 had a homozygous c.740_741del, p.(Val247Glufs*21) variant and showed profound intellectual disability, hepatomegaly, a simplified gyral pattern, and a thin corpus callosum without congenital dysmorphic features. Patient 2 had a compound heterozygous nonsense c.2124_2125del, p.(Phe709*) variant and splice site c.1188+2dup variant. RNA analysis revealed that the c.1188+2dup variant caused exon 13 skipping, leading to a frameshift (p.Ala406Ser*6). In vitro transcription analysis using minigene system suggested that mRNA transcribed from mutant allele may be degraded by nonsense-mediated mRNA decay system. He exhibited diverse manifestations, including growth defects, muscle hypotonia, respiratory distress, arthrogryposis, insulin-dependent diabetes mellitus, sensorineural hearing loss, facial dysmorphism, and various brain abnormalities, including cerebral atrophy, hypomyelination, and cerebellar hypoplasia. Here, we review previous literatures and discuss the phenotypic diversity of SMPD4-related disorders.
中文翻译:
两例双等位基因 SMPD4 变异伴脑结构异常的新病例
鞘磷脂磷酸二酯酶 4 ( SMPD4 ) 编码 Mg 2+依赖性中性鞘磷脂酶家族的成员,该家族催化鞘磷脂磷酸二酯键水解形成磷酸胆碱和神经酰胺。最近的研究表明,SMPD4的双等位基因功能丧失变异会导致综合征性神经发育障碍,其特征是小头畸形、先天性关节弯曲和大脑结构异常。在这项研究中,利用外显子组测序 (ES) 在两名患有发育迟缓、小头畸形、癫痫发作和脑结构异常的独立患者中鉴定出三种新的功能丧失型SMPD4变异体。患者 1 具有纯合 c.740_741del, p.(Val247Glufs*21) 变异,并表现出严重的智力障碍、肝肿大、简化的脑回模式和薄的胼胝体,但没有先天性畸形特征。患者 2 具有复合杂合无义 c.2124_2125del、p.(Phe709*) 变体和剪接位点 c.1188+2dup 变体。RNA 分析显示,c.1188+2dup 变体导致外显子 13 跳跃,导致移码 (p.Ala406Ser*6)。使用小基因系统进行的体外转录分析表明,从突变等位基因转录的 mRNA 可能会被无义介导的 mRNA 降解系统降解。他表现出多种表现,包括生长缺陷、肌张力低下、呼吸窘迫、关节挛缩、胰岛素依赖型糖尿病、感音神经性听力损失、面部畸形以及各种脑部异常,包括脑萎缩、髓鞘形成不足和小脑发育不全。在这里,我们回顾了以前的文献并讨论了SMPD4相关疾病的表型多样性。


























 京公网安备 11010802027423号
京公网安备 11010802027423号